
Today we present a paper from the team of Professor Charlotte M. Deane at the University of Oxford. Many studies have predicted that the integration of machine learning techniques into the development of small molecule therapeutics will help to achieve a real leap forward in drug discovery. However, despite advances in algorithms and novel architectures, improvements in outcomes have not always been significant. In this paper, the authors suggest that more attention should be paid to the data used to train and benchmark these models, which is more likely to drive future improvements. At the same time, the authors explore avenues for future research and strategies to address these data challenges.

Small molecule drugs, which account for 90 per cent of the world's approved drugs, are widely used in the pharmaceutical field because of their small molecular weight, which allows them to penetrate cell membranes and act directly on protein or RNA targets. However, the complexity of the small molecule drug development process and the high costs associated with failures in the preclinical and clinical phases have led to a halving of the number of approved drugs per $1 billion of R&D investment every nine years from 1950 to 2012, known as the ‘reverse Moore's law’.
Despite the success of machine learning (ML) in areas such as computer vision, natural language processing, and protein structure prediction, its application to small-molecule drug discovery has been poor. Studies have shown that these methods perform poorly when dealing with out-of-distribution data and struggle to maintain consistent performance on different benchmark tests. In addition, for known targets or regions of chemical space, simple methods or human experts may be sufficient, and complex ML methods are redundant in this context.The main applications of ML in small molecule drug discovery include molecular design, synthetic pathway prediction, molecular docking, and property prediction.
In a related field, AlphaFold 2 has achieved great success in protein structure prediction, which sheds light on small-molecule drug discovery. However, small-molecule drug discovery has not yet seen a similar ‘breakthrough moment’ and has only seen incremental improvements. This article analyses the challenges facing the field of small molecule drug discovery and discusses the direction of improvement in algorithms, data quality and quantity, and validation methods in order to achieve significant progress.
Impact of algorithm selection
The development of ML methods typically follows a pattern of proposing new architectures, training them with commonly used training sets, and testing them on commonly used benchmarks, which usually leads to only incremental improvements. In the case of CASF-2016, USPTO-50k, and HIV datasets, for example, no breakthroughs similar to AlphaFold 2 have occurred, although there have been improvements on USPTO-50k.
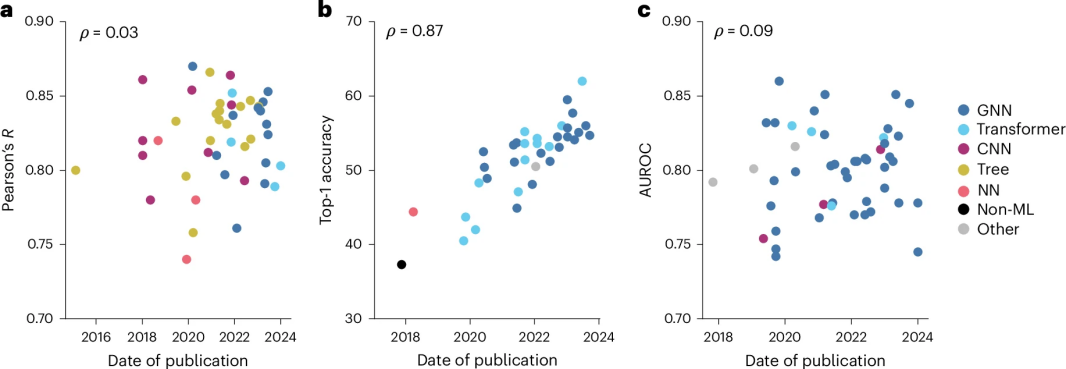
Advanced ML methods from other fields such as Transformer and Graph Neural Networks (GNN) have been used with limited effectiveness in small molecule drug discovery, as shown in Fig. 1.GNN can directly encode the molecular structures of compounds and targets, but it has limited effect on improving the accuracy and generality of small molecule methods. Extended GNNs do not significantly improve the actual results despite the symmetry of the 3D structure.
Diffusion models have been successful in generating images in other fields, but have failed to generate physically plausible molecules or effective protein interactions in the small molecule field, although accuracy has improved. Other new approaches such as coherence modelling and flow matching have also struggled to achieve breakthroughs on their own, and future advances will need to rely on a combination of algorithms, architectures, data quality and quantity.
Data Volume Issues
Scarcity of small molecule data
ML methods typically require large amounts of data for effective modelling, while biological and chemical data are expensive to generate and difficult to automate, resulting in far less small molecule data than image or text data. Compared to the billions or even trillions of data required for large model training, the current amount of publicly available data in the small molecule domain is only in the tens to hundreds of thousands. In addition, much of the data is retained by companies for intellectual property reasons, resulting in public data that relies primarily on academia or a small amount of proprietary data, such as KIBA, USPTO, and PDBBind. Addressing data scarcity is critical to advancing small molecule ML research.
Structural data
Docking algorithms, structure generation methods and scoring functions for small molecule ML methods rely on small molecule protein complex structure data, but currently available datasets (e.g., PDBBind) contain only 19,443 protein-ligand complexes, limiting model performance improvement. It is difficult to significantly increase the amount or diversity of data in the short term, so the utilisation of available structural data needs to be optimised. Current solutions such as data augmentation, which uses cross-docked conformations with small differences to crystalline ligands, are used to train generators and conformational classifiers. Additionally, synthetic conformations and synthetic pocket data generated based on the frequency of PDBBind interactions can also expand the dataset and improve accuracy. Self-distillation and physics-based pre-training can also improve prediction accuracy, and multiple methods may be integrated in the future to further enhance model performance.
Negative data
In small molecule drug discovery, negative data (e.g., ineffective binding or synthetic failures) are rarely made publicly available, often due to publication bias caused by recording only positive results. This phenomenon leads to an imbalance in the proportion of positives and negatives in the dataset, affecting the classification accuracy of ML models including, for example, yield prediction. Pharmaceutical companies sometimes need to reduce the weight of negative data or increase the sampling of positive data, but these data are usually not publicly available.
Strict publication guidelines, synthetic negative data, and crowdsourced data (e.g., the defunct Dark Response Database) have been suggested to address this issue, but may have limited effect. Another way to increase the amount of negative data is to extract complex data from papers and experimental results, with tools such as ChemDataExtractor, DECIMER.ai and Nougat automatically extracting information from the scientific literature.
Novel Data Sources
Changes in the field of drug discovery have led to new ways to increase data. For example, crowdsourced drug discovery enables drug development through an open and collaborative approach, allowing researchers around the world to propose compounds or ideas based on existing experimental data, unlocking a vast amount of data for ML to utilise. the COVID Moonshot project is an example of crowdsourced drug discovery, providing access to 470 crystal structures, IC50 test data on more than 2,000 compounds, and more than 3,000 synthetic compounds.
In addition, federated learning techniques in drug discovery allow for the sharing of private data without transferring data, such as the MELLODDY project and the Effiris project, which have expanded the domain of model applicability by 10 per cent and 83 per cent, respectively, through federated learning. These approaches help to increase the amount of training data for ML models, but the quality of the data is equally important.
Data quality control
Data bias
ML models learn trends from training data, but may learn biases that do not match reality and affect model accuracy. This is particularly common in small molecule research, where bias is caused by the fact that the data comes from many different sources and the sampling is biased towards a particular chemical space. Data biases also include ‘generalisation bias’, such as virtual screening tools that rely only on ligand features rather than protein-ligand interaction patterns, which affects the ability to generalise the model. To address this issue, methods such as asymmetric validation embedding can be used to generate more stringent training-validation splits to avoid models memorising ligand features. With the attention paid to the bias problem in the field, learning or application domain adaptation to avoid invalid bias becomes a key strategy.
Data Noise
A major limitation of small molecule data is the high amount of noise, usually caused by merging data from different sources. For example, merging IC50 data from different experimental conditions to train a QSAR model can lead to increased uncertainty and inflate model accuracy metrics. In addition, uncertainty is unlabelled in many datasets and needs to be estimated relying on data type or experimental approximations.
Another source of noise is false-positive results, such as pan-agent interfering compounds (PAINs), which are susceptible to non-specific binding to multiple targets, leading to false positives. Existing methods can detect PAINs by molecular substructural markers, but datasets are often not examined for these markers. In addition, predictions based on the frequency of PAINs target matches show a high noise problem. To reduce noise, easy-to-use data cleaning processes need to be established and collaboration between experimental and computational studies needs to be facilitated to optimise data quality. Data quality management and validation are key aspects in small molecule ML development.
Method Validation
Uncovering Performance Enhancement
Validation of small molecule ML methods is usually performed by using test sets homologous to the training set or standard benchmark sets, and the enhancements are mostly incremental, making it difficult to demonstrate their unique benefits in drug discovery. Baseline and ablation tests can help assess the actual effect of increased model features and complexity. For example, simple decision tree scoring functions based only on ligand or protein features are comparable in accuracy to more complex methods, suggesting that many of the performance gains may stem from dataset bias rather than generalised physical characteristics. Ablation tests should be more general in order to provide validation of effectiveness as data choices diversify.Ablation tests for AlphaFold 2 show that multiple innovative features, rather than a single factor, combine to improve model accuracy. While these methods reveal sources of improvement, their applicability to drug discovery requires further validation.
Robustness testing
Methods in this field are typically evaluated on training data, but the type of data in real-world drug discovery may be different and not adequately considered. For example, ML-based protein-ligand docking is often used to ‘re-dock’ to test known structures, leading to over-optimism about actual accuracy. When evaluated with more realistic test sets, the accuracy is significantly reduced. Similarly, inverse synthesis methods are often tested on single-step predictions, but in practice they are often combined with path search algorithms, where single-step accuracy does not reflect the success rate of the complete pathway. The small size of existing benchmark datasets makes it difficult to adequately evaluate the models. More rigorous validation and benchmarking is needed to more closely match real-world needs, and online leaderboards and public ‘blind’ prediction competitions (e.g., D3R, CACHE) can provide a fairer assessment of models. Such robust validation is essential to ensure the effectiveness of ML in drug discovery in practice.
Conclusions and future perspectives
Revolutionising small molecule therapeutic development through ML relies not only on the sophistication of the algorithms, but also on the quality, diversity and quantity of training and validation data. Key current challenges are that ML models generalise poorly to data outside the training distribution and are prone to learning confounding trends and biases in the dataset. These issues suggest that a refocus on data and validation is needed to realise the full potential of ML algorithms.
This paper analyses the limitations of public data and its impact on model training and validation, and explores future directions. To cope with these issues, data augmentation, data crawling, crowdsourced data and federated learning are suggested to increase the amount of data and to deal with confounding factors in the data such as PAINS compounds, experimental condition noise and data bias. In addition, strict training/validation data partitioning needs to be used to avoid models learning only dataset bias. With these improvements, ML-driven small molecule drug development remains promising, but realising this potential requires a balanced focus on all elements of model development, including ML architecture, data quality and validation methods.
Reference : Durant G, Boyles F, Birchall K, et al. The future of machine learning for small-molecule drug discovery will be driven by data[J]. Nature Computational Science, 2024: 1-9.







Post comments