
by Juntendo University Research Promotion Center
Credit: npj Genomic Medicine (2024). DOI: 10.1038/s41525-024-00429-5
Mitochondrial diseases are among the most prevalent hereditary metabolic disorders, known to occur in 1 out of every 5,000 births. Single nucleotide variations, indels, and structural variations are known to cause these disorders.
While many arise from single nucleotide variations, indels, or structural variants, some forms are also triggered by repeat expansions in nuclear genes affecting mitochondrial function, which can result in severe mitochondrial dysfunction. These diseases often impact the central nervous system (CNS), and mitochondrial encephalopathies represent a subset characterized by prominent CNS lesions.
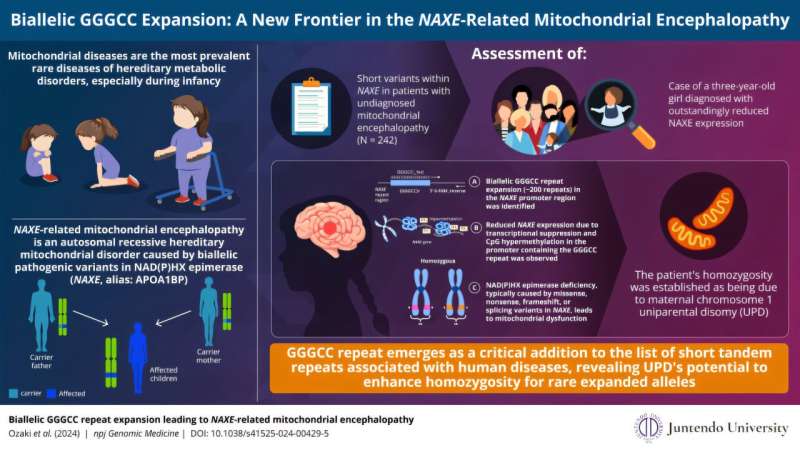
In a new study published in npj Genomic Medicine on October 25, 2024, a research team led by Professor Yasushi Okazaki, along with Dr. Kei Murayama from the Division of Diagnostics and Therapeutics of Intractable Diseases at the Intractable Disease Research Center in Juntendo University, Japan, used advanced techniques to identify a biallelic GGGCC repeat expansion that caused NAXE-related mitochondrial encephalopathy in a 3-year-old girl.
An autosomal recessive hereditary disorder, NAXE-related mitochondrial encephalopathy is characterized by biallelic pathogenic variants in NAD(P)HX epimerase, which leads to accumulation of a toxic metabolite that damages mitochondrial function resulting from a deficiency in the enzyme.
Prof. Okazaki, who led this study, explains, "As part of our strategy to achieve molecular diagnoses on 2,932 undiagnosed patients with mitochondrial diseases in Japan, we conducted RNA sequencing of samples from fibroblasts obtained from 400 patients, in addition to genomic sequencing."
Of these patients, 303 were biochemically confirmed to have mitochondrial disorders and the team performed bioinformatics analysis to isolate individuals with marked dysregulation of candidate causative genes. It is then that the case of a 3-year-old girl with severely reduced NAXE gene expression was discovered.
This patient did not report a familial history of mitochondrial, rare, or neurodegenerative diseases, but clinically presented with truncal instability while sitting, mild psychomotor developmental delay, seizure, and difficulty hearing in the right ear. Using long read sequencing, the team found almost 200 repeats of GGGCC in the patient's NAXE promoter region, compared to just three repeats in the reference sequence.
The team analyzed the unspliced NAXE transcripts from this patient to examine the molecular mechanism underlying the severe reduction in NAXE transcripts and protein. They found that transcriptional suppression occurred due to hypermethylation of the repeat regions in the NAXE promoter regions.
In addition, they observed hypermethylation on the repeat regions in one of the alleles in the patient's mother. The mother's allele with the normal range of repeats were not hypermethylated, however. Genetic analysis of the patient's sample demonstrated that the homozygosity of the repeat expansion is due to the maternal chromosome 1 uniparental disomy (UPD), an abnormal condition where both members of a chromosome pair are inherited from one parent.
Prof. Okazaki says, "We also performed amplicon long read sequencing." Pathogenic repeat expansions often cause cytotoxicity resulting from translated peptide accumulation. However, in this case of GGGCC repeat expansion in NAXE, NAXE mRNA was extremely decreased, suggesting that it could minimize the impacts of such mechanisms.
Prof. Okazaki points out a few limitations in their study that could open up scope for further research. "We found only a single case of repeat expansion in the pedigree, warranting further investigations in more cohorts.
"Our experiments to identify pathogenic GGGCC repeat expansions involved polymerase chain reaction (PCR)-based techniques, so we could have overlooked PCR-resistant structural variants of such repeats. Also, we have not tried to measure the toxic metabolites like cyclic NADHX, which is a direct consequence of the dysfunction of NAD(P)HX epimerase encoded by NAXE."
Overall, the study emphasizes the importance of advanced sequencing technologies in identifying previously unknown genetic causes of mitochondrial diseases, suggesting that UPD, which occurs in approximately 0.05% of live births, could play a significant role in homozygosity for rare repeat expansions and help diagnose patients.
Hopefully, further research can help reveal the pathogenicity of other potential NAXE variants, allowing researchers to investigate the occurrence of GGGCC expansions in larger patient cohorts.
More information: Kokoro Ozaki et al, Biallelic GGGCC repeat expansion leading to NAXE-related mitochondrial encephalopathy, npj Genomic Medicine (2024). DOI: 10.1038/s41525-024-00429-5
Provided by Juntendo University Research Promotion Center




Post comments