
by Sarah C. P. Williams, Gladstone Institutes
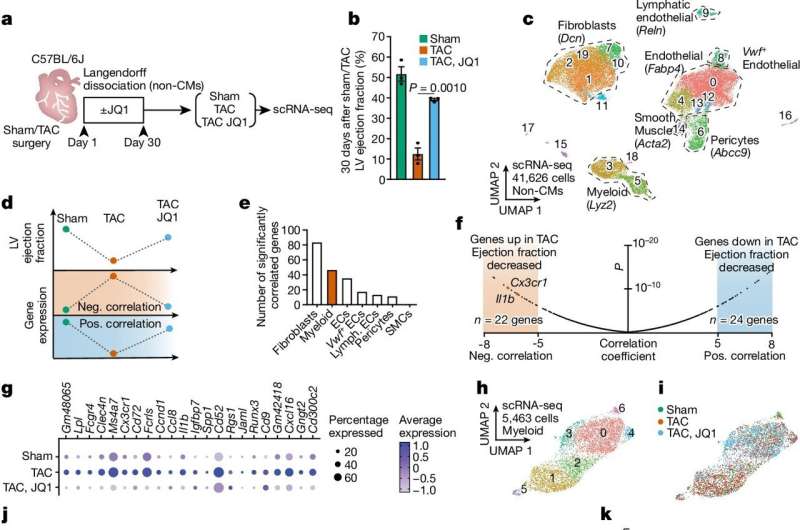
Stress-activated Cx3cr1+ macrophages contribute to heart failure pathogenesis. Credit: Nature (2024). DOI: 10.1038/s41586-024-08085-6
If you cut your arm or undergo surgery, scarring can be a good thing; the scar tissue produced by cells called fibroblasts helps you heal. In most organs of the body, however, the accumulation of scarring (called fibrosis) is a sign of chronic disease and aging. Slowing or stopping fibrosis can help treat heart, liver, kidney, and lung diseases. Yet fibrosis of these organs remains a deadly disease with limited treatments.
Now, scientists at Gladstone Institutes have worked out exactly how immune cells trigger scar-forming fibroblasts to become activated and cause fibrosis in the heart, and likely in other organs as well. Blocking the molecular signals between inflammation-causing immune cells and fibroblasts, they showed, can prevent this fibrosis. And, by chance, some of the signals are already targeted by existing drugs, which could potentially be repurposed for heart disease.
"The crosstalk between the immune system and fibroblasts is even more complex than we expected," says Gladstone Assistant Investigator Michael Alexanian, Ph.D., the corresponding author of the new study published in Nature. "But now that we've begun to map it out, we may be able to develop therapies that target both inflammation and fibrosis."
"Inflammation and fibrosis both play incredibly important roles in the worsening of heart failure, which affects 25 million people worldwide, and many other fibrotic diseases," adds Deepak Srivastava, MD, president and senior investigator at Gladstone and senior author of the study. "Discovering new ways to stop these processes could help millions of people."
Heart disease changes both immune cells and fibroblasts
Scientists have long known that both inflammation and fibrosis occur together in diseased organs and with normal aging. Fibrosis in the heart, liver, kidney, and lungs causes unhealthy thickening and stiffening of the organs, which impedes their functions and contributes to organ failure. But the precise role of inflammation, and how it directly causes fibrosis, has not been fully understood.
In the new work, the group used mice to probe how immune cells, in addition to fibroblasts, change during heart disease. They also treated the mice with a drug called a BET inhibitor, which they previously showed prevents fibrosis in animals but has many side effects in other tissues. Their goal was to discern what happens to the cells when heart disease is treated and glean insights that could be used in developing more targeted therapies for humans.
"A number of pro-inflammatory genes were activated in immune cells during heart failure and then repressed during treatment," says Arun Padmanabhan, MD, Ph.D., cardiologist, co-first author of the new paper, and former postdoctoral scholar in Srivastava's lab at Gladstone, who is now an investigator at UC San Francisco (UCSF). "So, it was immediately clear that with this drug, we were shutting down inflammation as well as fibrosis."
A signal from immune cells to fibroblasts
Then, through a series of experiments on both the immune cells and fibroblasts, the researchers discovered new molecular connections between inflammation and fibrosis. They detailed a cascade of events that begins with macrophages, immune cells that play a central role in triggering inflammation.
In 2021, the same team had discovered that in fibroblasts, turning on a gene called MEOX1 triggers fibrosis by activating the cells. However, they didn't know how stress in the heart was sensed by the fibroblasts to become activated.
In the new work, the scientists discovered that when the heart is in a diseased state, the regulatory gene Brd4 is turned on in macrophages, and it makes the immune cells produce IL-1β, a signaling molecule. When it interacts with neighboring fibroblasts, the IL-1β protein activates the "master switch" MEOX1 in fibroblasts—which researchers had already linked to fibrosis.
"We were able to pinpoint exactly where the inflammation starts in macrophages, and how that affects fibroblasts," says Padmanabhan. "Understanding this entire chain of events will allow us to intervene earlier in the process from multiple angles and find better ways to prevent fibrosis in patients."
In fact, the researchers showed that, in mice, blocking different steps of the macrophage-fibrosis pathway—including Brd4 and IL-1β—can help ease heart fibrosis in animals with heart disease.
In addition, because the IL-1β protein has other roles in driving inflammation throughout the body, drugs have already been developed to stop its activity in diseases including cancer, rheumatoid arthritis, and diabetes. But determining whether these drugs influence fibrosis in the heart or other organs will require additional studies.
"Now that we know the signals involved, we can develop targeted drugs that stop fibrosis without blocking the positive effects of the immune system, such as fighting infection," says Alexanian, who is also a professor in the Department of Pediatrics at UCSF.
More information: Michael Alexanian et al, Chromatin remodelling drives immune cell–fibroblast communication in heart failure, Nature (2024). DOI: 10.1038/s41586-024-08085-6
Journal information: Nature
Provided by Gladstone Institutes





Post comments