
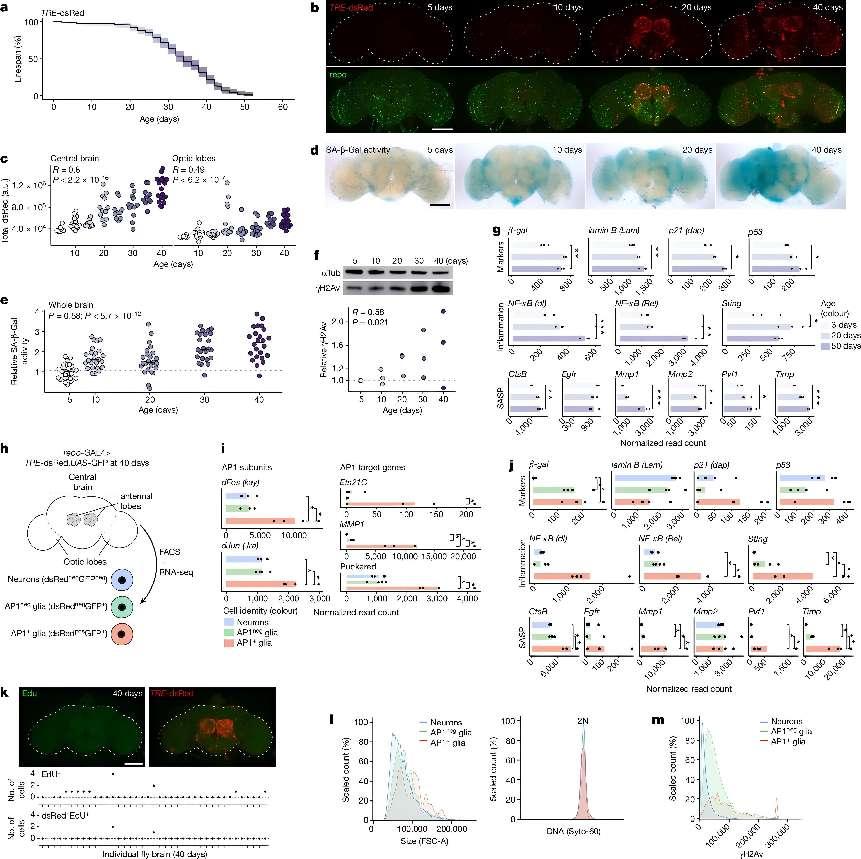
AP1 glia appear with age and have a senescent phenotype.+a, Lifespan of genomic AP1 reporter line (TRE-dsRed; n = 200 flies). b, Representative images of fly brains showing age-onset AP1 activity (dsRed; top) is mostly in glial cells (repo; bottom). c, Quantification of dsRed intensity shows that AP1 activity is higher the central brain (left) versus optic lobes (right) (TRE-dsRed; n = 93 brains). See Extended Data Fig. 1a–c for high-magnification images and quantification of dsRed colocalization with glial versus neuronal markers. a.u., arbitrary units. d,e, Representative images showing SA-β-Gal activity increases with age (d) with quantification (e) (TRE-dsRed; n = 119 brains). f, Brain γH2Av levels increase with age (TRE-dsRed; n = 8 brains per replicate). For gel source data, see Supplementary Fig. 1a. g, Bulk RNA-seq of brains shows that senescence-associated genes increase with age (w1118; n = 20 brains per replicate). h, Cells were FACS-isolated from 40-day-old brains for bulk RNA-seq (repo-GAL4 > TRE-dsRed;UAS-GFP; n = 500 cells per replicate). i,j, Expression of AP1 subunits (dFos, dJun), AP1-target genes (i) and senescence-associated genes (j) is highest in AP1 glia. See Supplementary Data 1 for differential expression genes. k, Most AP1 glia are non-dividing by EdU labelling at 40 days (TRE-dsRed; n = 39 brains). l, Analysis of live FACS-isolated cells from 40-day-old brains shows that AP1 glia are larger (left) with normal DNA content (right) (n = 4,748 neurons, n = 14,326 AP1+++neg glia, n = 490 AP1 glia). m, Distribution of γH2Av staining in fixed FACS-isolated cells from 40-day-old brains (n = 8,609 neurons, n = 845 AP1+neg glia, n = 302 AP1 glia). For all bar graphs, data shown are means. Each point in a microscopy experiment represents one brain; in immunoblot or bulk RNA-seq experiments it represents one biological replicate. All data were collected from two or three independent experiments. Pearson’s correlation (c,e,f). Precise n and P values are in the Source Data. *P-adjusted < 0.05 for sequencing data; ***P < 0.001; **P < 0.01, *P < 0.05 for all other data. All scale bars, 100 μm.+
The aging process is characterized by a gradual decline in physiological functions and an increased susceptibility to age-related diseases. While some aspects of aging, such as physical frailty and cognitive decline, are well understood, the underlying mechanisms that drive these changes remain elusive. One emerging area of research focuses on cellular senescence, a state of irreversible growth arrest and increased secretion of inflammatory factors. Studies in both humans and model organisms have highlighted the potential role of senescent cells in driving aging-related pathologies. However, the precise mechanisms by which senescent cells contribute to aging and their interplay with other cellular processes remain largely unknown.
The intricate relationship between cellular senescence, mitochondrial dysfunction, and lipid accumulation emerges as a key area of investigation in understanding the aging process. Using the fruit fly, Drosophila melanogaster, as a powerful model organism, Byrns et al. have shed light on a novel mechanism by which senescent glial cells, a subset of cells that undergo a state of permanent growth arrest and secrete inflammatory factors, contribute to the aging process. The study published in Nature revealed that senescent glial cells, which first appeared in the degenerating antennal lobes of the fly brain and later spread to the optic lobes, not only exhibited hallmarks of senescence such as increased SASP gene expression and DNA damage but also played a pivotal role in triggering lipid accumulation in non-senescent glial cells. This accumulation, driven by factors produced by senescent cells, led to the formation of lipid droplets, which were associated with both protective and detrimental effects in the brain.
More specifically, the study began by leveraging a transgenic line expressing a fluorescent marker (dsRed) under the control of the Activator protein 1 (AP1) binding motif. This allowed the researchers to visualize and track the appearance of senescent-like glial cells throughout the fly's lifespan. They observed a distinct pattern of accumulation, with these cells first appearing in the antennal lobes, known to degenerate with age, and later spreading to the optic lobes.
The researchers characterized the senescent glial cells using RNA sequencing and found that they exhibited hallmarks of senescence, including increased expression of senescence-associated secretory phenotype (SASP) genes, cell cycle arrest, increased metabolic activity, and DNA damage. They also discovered a correlation between the appearance of these cells and declining mitochondrial function in neurons. Through a targeted RNA interference screen, they found that knocking down specific mitochondrial genes in neurons led to the activation of AP1 and the subsequent appearance of senescent glial cells. This suggested that neuronal mitochondrial dysfunction could be a key trigger for glial senescence.
To assess the impact of senescent glial cells on fly lifespan and healthspan, the researchers used an inducible system to block AP1 activity in glial cells. They found that continuous blockade of AP1 resulted in a significant reduction in lifespan, while intermittent blockade (once or three times a week) had a more modest effect. However, blocking AP1 for just one day per week extended median and maximum lifespan, improved locomotor activity, and reduced the expression of senescence markers in the brain. This suggested that a mild reduction in AP1 activity could mitigate some of the negative effects of senescence.
Further investigation revealed that senescent glial cells played a role in promoting lipid accumulation in non-senescent glial cells. The researchers observed an increase in lipid droplets (LDs) in non-senescent glial cells with age, and blocking AP1 activity significantly reduced the number and size of these LDs. They also found that senescent glial cells expressed genes involved in lipid synthesis and contained high levels of free fatty acids (FFAs), which could potentially be transferred to non-senescent glial cells.
To validate their findings in a different system, the researchers conducted experiments in human fibroblast cell cultures. They induced senescence in these cells and found that the conditioned medium from senescent cells promoted LD formation in naive cells, while the medium from cells with reduced AP1 activity had the opposite effect. This supported the idea that senescent cells can influence lipid accumulation in non-senescent cells.
While targeting senescent glial cells appeared to extend lifespan and healthspan, the researchers also observed an increase in oxidative stress markers in the brains of flies with reduced AP1 activity. This suggested that while senescent cells may promote aging-related pathologies, their removal may also lead to increased vulnerability to oxidative damage.
This study provides compelling evidence that senescent glial cells play a significant role in the aging brain and contribute to both beneficial and detrimental effects. The identification of neuronal mitochondrial dysfunction as a key trigger for glial senescence opens up new avenues for research into aging and potential therapeutic strategies. Further investigation is needed to understand the mechanisms by which senescent glial cells promote lipid accumulation and the potential consequences of this process. Additionally, exploring ways to mitigate oxidative stress in the brain while targeting senescent cells could lead to more effective anti-aging interventions.
Reference:






Post comments