
by Ben Leach, Children's Hospital of Philadelphia
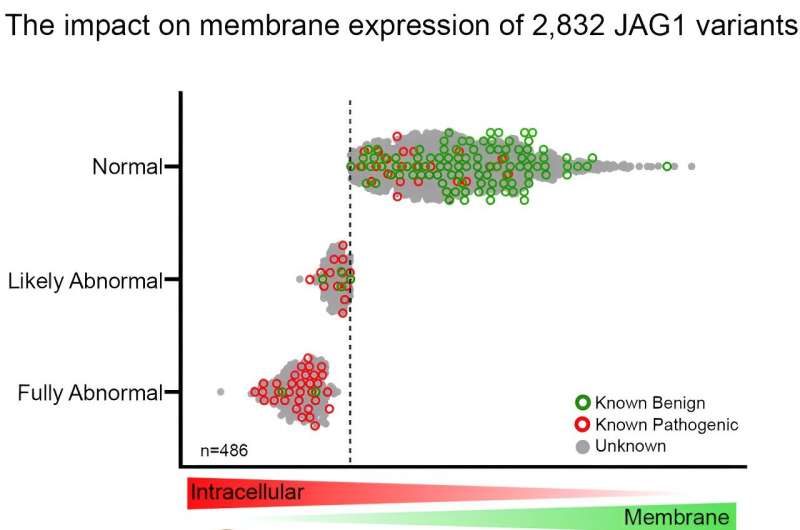
Credit: The American Journal of Human Genetics (2024). DOI: 10.1016/j.ajhg.2024.06.011
Researchers from Children's Hospital of Philadelphia (CHOP) and the Perelman School of Medicine of the University of Pennsylvania (Penn Medicine) developed data designed to improve the diagnosis for Alagille syndrome, a complex inherited liver disorder. These findings will help to reduce the Variant of Uncertain Significance (VOUS) classification for missense variants, protein-altering genetic changes, which has implications for other diseases. The findings were published today by the American Journal of Human Genetics.
Alagille syndrome—also known as Alagille-Watson syndrome, syndromic bile duct paucity and arteriohepatic dysplasia—is an inherited liver disorder that also affects the heart, eyes, bones, kidneys, vasculature and other organs, with a wide range of severity. Alagille syndrome is predominately caused by variants in the Jagged1 (JAG1) gene, which helps make a protein that controls how cells grow and develop into different types of tissues and organs when functioning properly.
Many protein-shortening genetic changes can be easily identified as disease-causing and lead to proper diagnosis of Alagille syndrome, allowing children to receive an appropriate course of treatment. However, up to 85% of reported JAG1 missense variants—mutations where one amino acid is replaced by a different one that is not typically in that position in the gene—have uncertain or conflicting classifications, meaning that patients with Alagille syndrome could be missed.
"We wanted to focus on the exons where these missense variants were most likely to occur and parse out variants that could be compared to ones we know are responsible for disease," said the study's first author Melissa Gilbert, Ph.D., Research Assistant Professor of Pathology and Laboratory Medicine at Penn Medicine and CHOP. "Using a variety of methods, we developed a high-throughput assay and were able to reduce uncertainty when it comes to classifying these missense variants and improving our ability to provide a more accurate diagnosis of Alagille syndrome."
Every gene contains exons, which are the coding regions that provide instructions for making specific proteins. In this study, researchers generated a library of 2,832 JAG1 nucleotide variants within exons 1 through 7, a region of the gene with a high number of reported missense variants.
Using a high-throughput assay to measure JAG1 membrane expression and calibrating using a separate set of control variants, the researchers characterized 486 variants as functionally abnormal, of which 439 were missense variants.
This information was then compared with 144 uncertain variants reported in patients undergoing clinical or research testing. Of those variants, 27 had functionally abnormal membrane expression of JAG1, and as a result of these findings, 26 were reclassified as likely pathogenic or causing Alagille syndrome.
"The method we used to re-characterize these variants has implications for other genetic drivers of rare and complex diseases," said senior study author Nancy B. Spinner, Ph.D., Chief of the Division of Genomic Diagnostics at CHOP. "In the immediate future, we hope that this dataset can provide genetic counselors and clinicians with an enhanced ability of classifying these variants when they are first detected in patients with Alagille syndrome and reduce the uncertainty and anxiety faced by families with patients faced with this challenging disease."
The researchers' future goals include expanding this work to solve all variants in JAG1, expanding to other disease genes, and developing protocols to embed this data into the interpretation of variants in the clinical diagnostic lab at CHOP. Solving VOUS across all medically significant genes is a stated priority of the National Human Genome Research Institute.
More information: Melissa A. Gilbert et al, Functional characterization of 2,832 JAG1 variants supports reclassification for Alagille syndrome and improves guidance for clinical variant interpretation, The American Journal of Human Genetics (2024). DOI: 10.1016/j.ajhg.2024.06.011
Journal information: American Journal of Human Genetics
Provided by Children's Hospital of Philadelphia





Post comments