
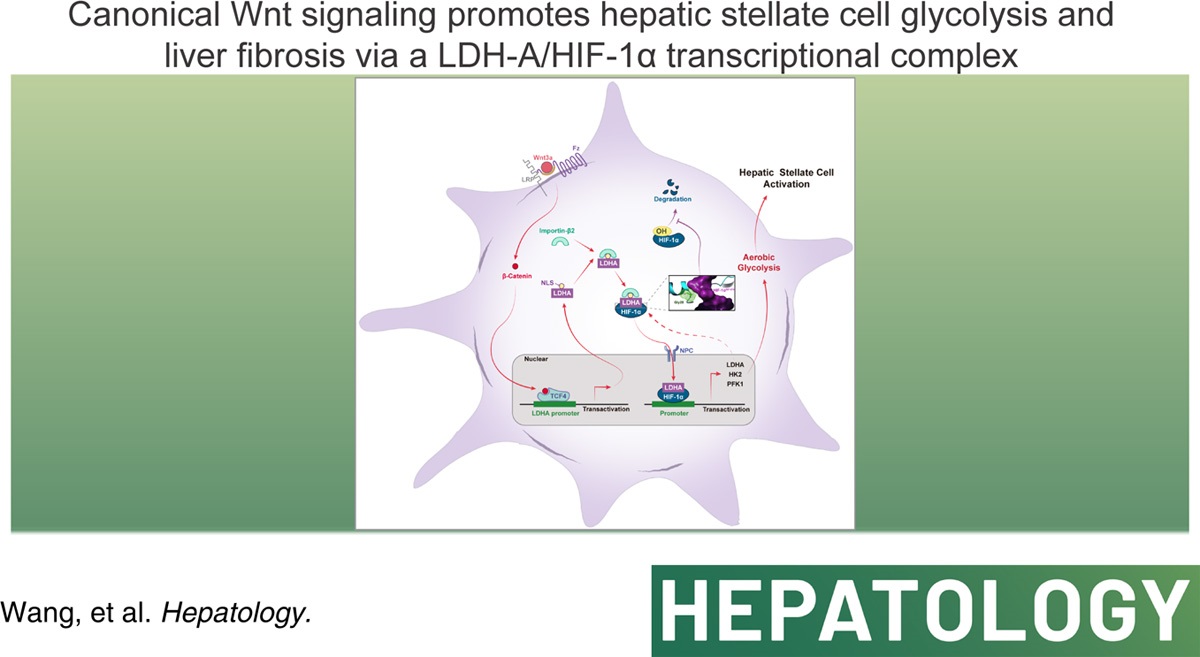
Credit:Canonical Wnt signaling promotes HSC glycolysis and liver fibrosis through an LDH-A/HIF-1α transcriptional complex.
This comprehensive study conducted by Wang et al. delved into the intricate relationship between canonical Wnt signaling, hepatic stellate cell (HSC) activation, and liver fibrosis. The authors utilized a multifaceted approach encompassing cellular and molecular biology, computational simulations, and animal models to elucidate the mechanisms by which Wnt signaling drived HSC glycolysis and subsequent liver fibrosis.
The study began by demonstrating that canonical Wnt signaling, mediated by β-catenin, significantly enhanced aerobic glycolysis in HSCs. This metabolic shift was characterized by increased glucose uptake and consumption, elevated lactate production, and suppression of oxidative phosphorylation. The authors employed RNA sequencing and metabolic flux analysis to identify key glycolytic enzymes, including hexokinase 2 (HK2), phosphofructokinase 1 (PFK1), and lactate dehydrogenase A (LDH-A), as downstream targets of Wnt/β-catenin signaling.
Further investigation revealed that LDH-A played a central role in Wnt/β-catenin-induced glycolysis. LDH-A was rapidly upregulated upon Wnt3a stimulation through a TCF4-dependent transcriptional mechanism. This finding resolved the ambiguity surrounding the relationship between β-catenin activation and LDH-A expression, establishing LDH-A as a direct target gene of the β-catenin/TCF4 complex.
Surprisingly, LDH-A was not only a glycolytic enzyme but also exhibited nuclear translocation upon Wnt activation. This phenomenon was facilitated by importin β2 and a noncanonical nuclear localization signal (NLS) within LDH-A. The nuclear accumulation of LDH-A was crucial for its interaction with hypoxia-inducible factor-1α (HIF-1α), a master regulator of glycolysis.
Then, the study uncovered a direct physical interaction between LDH-A and HIF-1α, leading to the stabilization of HIF-1α and enhanced transactivation of glycolytic genes. Molecular simulations and mutational analyses identified the Gly28 residue of LDH-A as the key site for binding to HIF-1α. This interaction prevented the hydroxylation and subsequent proteasomal degradation of HIF-1α, thereby promoting its nuclear accumulation and transcriptional activity.
Furthermore, the authors demonstrated that LDH-A-mediated aerobic glycolysis was essential for Wnt/β-catenin-induced HSC activation. Inhibition of glycolysis or LDH-A expression abrogated Wnt3a-stimulated HSC proliferation, migration, contraction, and the expression of α-smooth muscle actin (α-SMA) and collagen 1α1 (COL1α1), hallmark markers of HSC activation.
The in vivo relevance of these findings was confirmed using a mouse model of liver fibrosis induced by carbon tetrachloride (CCl4). HSC-specific knockdown of LDH-A significantly mitigated liver fibrosis, while HSC-specific overexpression of LDH-A exacerbated the disease. Pharmacological inhibition of β-catenin with XAV939 also reduced HSC activation and liver fibrosis, an effect that was reversed by HSC-specific LDH-A overexpression.
Finally, the study demonstrated a positive correlation between β-catenin and LDH-A expression in human liver fibrosis samples. This clinical correlation reinforced the relevance of the identified signaling pathways and metabolic alterations in human liver fibrosis.
In summary, this study elucidates a critical role for canonical Wnt signaling and LDH-A in HSC metabolism and liver fibrosis. The identified signaling pathways and metabolic alterations offer valuable insights for the development of novel therapeutic strategies to combat this debilitating liver disease.
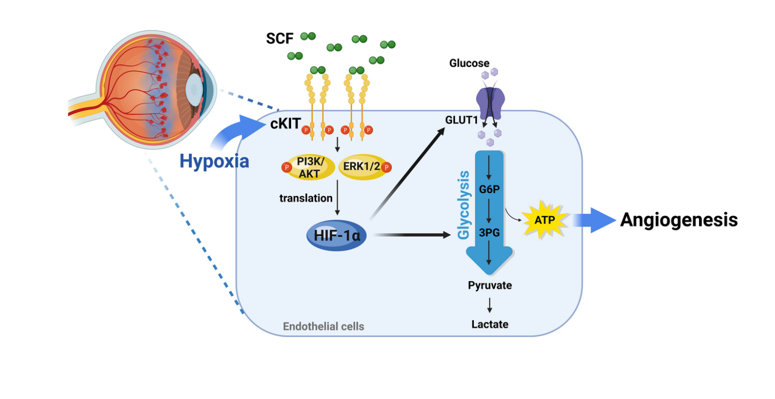
Credit:Stem cell factor and cKIT modulate endothelial glycolysis in hypoxia.
Another relevant study conducted by Jeong et al. explored the intricate interplay between hypoxia, angiogenesis, and endothelial metabolism, focusing on the role of stem cell factor (SCF) and its receptor, cKIT. The authors hypothesized that SCF/cKIT signaling played a crucial role in regulating endothelial glycolysis during hypoxia-driven angiogenesis, ultimately contributing to pathological neovascularization.
Hypoxia, characterized by a lack of oxygen supply, is a common feature of various pathological conditions, including ischemia, cancer, and retinopathy. Endothelial cells (ECs), the linings of blood vessels, respond to hypoxia by undergoing a metabolic shift from oxidative phosphorylation to glycolysis. This shift enables ECs to meet the increased energy demands associated with angiogenesis, the formation of new blood vessels. Glycolysis, an oxygen-independent metabolic pathway, provides ECs with a rapid source of ATP and metabolic intermediates necessary for cell proliferation, migration, and vessel formation.
The authors previously demonstrated that hypoxia induced cKIT expression in ECs and that SCF/cKIT signaling contributed to pathological neovascularization in ischemic tissues. Building upon this foundation, the current study investigated the role of SCF/cKIT signaling in regulating endothelial glycolysis during hypoxia-driven angiogenesis. As expected, SCF/cKIT signaling significantly increased glucose uptake, lactate production, and glycolytic capacity in human umbilical vein endothelial cells (HUVECs) under hypoxia. This effect was abolished by cKIT inhibition, confirming the involvement of cKIT in mediating SCF-induced glycolysis.
Mechanistically, SCF/cKIT signaling enhanced the expression of key glycolytic enzymes and glucose transporter 1 (GLUT1) in hypoxic ECs. This effect was also dependent on cKIT activation and could be reversed by inhibiting glycolysis or HIF-1α. In addition, SCF/cKIT signaling increased HIF-1α protein levels without altering its mRNA levels, indicating a post-transcriptional regulation. This effect was mediated by AKT and ERK1/2 signaling pathways, which phosphorylated the translation regulatory protein p70S6K and promoted HIF-1α translation.
To explore the role of glycolysis in angiogenesis promoted by SCF/cKIT Signaling, the authors employed inhibitors of glycolysis or HIF-1α, which significantly attenuated SCF-induced angiogenic activities in vitro, demonstrating the critical role of HIF-1α and glycolysis in SCF-mediated angiogenesis. In vivo, OIR mice exhibited increased expression of SCF, cKIT, HIF-1α, and glycolytic enzymes in the retina. Blocking SCF/cKIT signaling using anti-SCF antibodies or cKit mutant mice reduced the expression of HIF-1α and glycolytic enzymes and suppressed pathological neovascularization in OIR mice.
In conclusion, the studies by Wang et al. and Jeong et al. highlighted the pivotal role of metabolic reprogramming and HIF-1α signaling in liver fibrosis and pathological angiogenesis. Wang et al. demonstrated that canonical Wnt signaling promoted HSC glycolysis and liver fibrosis through β-catenin-mediated upregulation of LDH-A, which stabilized HIF-1α and enhanced glycolytic gene expression. The other study revealed that SCF/cKIT signaling regulated endothelial glycolysis and angiogenesis under hypoxia, with HIF-1α playing a central role in this process. These findings underscore the importance of targeting metabolic pathways and HIF-1α in the development of novel therapeutic strategies for liver fibrosis and angiogenesis-related diseases.
Reference:
Wang F, Chen L, Kong D, Zhang X, Xia S, Liang B, Li Y, Zhou Y, Zhang Z, Shao J, Zheng S, Zhang F. Canonical Wnt signaling promotes HSC glycolysis and liver fibrosis through an LDH-A/HIF-1α transcriptional complex. Hepatology. 2024 Mar 1;79(3):606-623.
Jeong H, Kim RI, Koo H, Choi YH, Kim M, Roh H, Park SG, Sung JH, Kim KL, Suh W. Stem cell factor and cKIT modulate endothelial glycolysis in hypoxia. Cardiovasc Res. 2024 May 29;120(7):745-755.





Post comments