
by University of Chicago

DNA, which has a double-helix structure, can have many genetic mutations and variations. Credit: NIH
A new statistical tool developed by researchers at the University of Chicago improves the ability to find genetic variants that cause disease. The tool, described in a new paper published January 26, 2024, in Nature Genetics, combines data from genome-wide association studies (GWAS) and predictions of genetic expression to limit the number of false positives and more accurately identify causal genes and variants for a disease.
GWAS is a commonly used approach to identify genes associated with a range of human traits, including most common diseases. Researchers compare genome sequences of a large group of people with a specific disease, for example, with another set of sequences from healthy individuals. The differences identified in the disease group could point to genetic variants that increase risk for that disease and warrant further study.
Most human diseases are not caused by a single genetic variation, however. Instead, they are the result of a complex interaction of multiple genes, environmental factors, and host of other variables. As a result, GWAS often identifies many variants across many regions in the genome that are associated with a disease.
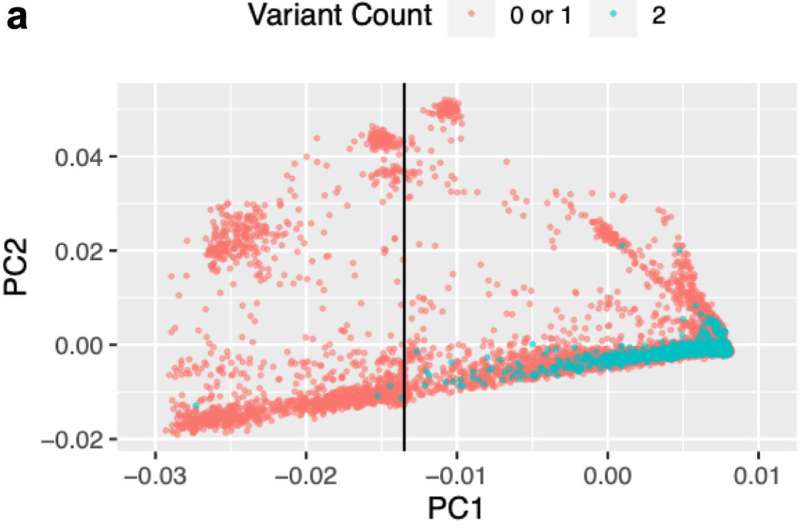
The limitation of GWAS, however, is that it only identifies association, not causality. In a typical genomic region, many variants are highly correlated with each other, due to a phenomenon called linkage disequilibrium. This is because DNA is passed from one generation to the next in entire blocks, not individual genes, so nearby variants tend to be correlated.
"You may have many genetic variants in a block that are all correlated with disease risk, but you don't know which one is actually the causal variant," said Xin He, Ph.D., Associate Professor of Human Genetics, and senior author of the new study. "That's the fundamental challenge of GWAS, that is, how we go from association to causality."
To make the problem even harder, most of the genetic variants are located in non-coding genomes, making their effects difficult to interpret. A common strategy to address these challenges is using gene expression levels. Expression quantitative trait loci, or eQTLs, are genetic variants associated with gene expression.
The rationale of using eQTL data is that if a variant associated with a disease is an eQTL of some gene X, then X is possibly the link between the variant and the disease. The problem with this reasoning, however, is that nearby variants and eQTLs of other genes can be correlated with the eQTL of the gene X while affecting the disease directly, leading to a false positive.
Many methods have been developed to nominate risk genes from GWAS using eQTL data, but they all suffer from this fundamental problem of confounding by nearby associations. In fact, existing methods can generate false positive genes more than 50% of the time.
In the new study, Prof. He and Matthew Stephens, Ph.D., the Ralph W. Gerard Professor and Chair of the Departments of Statistics and Professor of Human Genetics, developed a new method called causal-Transcriptome-wide Association studies, or cTWAS, that uses advanced statistical techniques to reduce false positive rates. Instead of focusing on just one gene at a time, the new cTWAS model accounts for multiple genes and variants. Using a Bayesian multiple regression model, it can weed out confounding genes and variants.
"If you look at one at a time, you'll have false positives, but if you look at all the nearby genes and variants together, you are much more likely to find the causal gene," He said.
The paper demonstrates the utility of this new technique by studying genetics of LDL cholesterol levels. As one example, existing eQTL methods nominated a gene involved in DNA repair, but the new cTWAS approach pointed at a different variant in the target gene of statin, a common drug used to treat high cholesterol. In total, cTWAS identified 35 putative causal genes of LDL, more than half of which have not been previously reported. These results point to new biological pathways and potential treatment targets for LDL.
The cTWAS software is now available to download from He's lab website. He hopes to continue working on it to extend its capabilities to incorporate other types of 'omics data, such as splicing and epigenetics, as well as using eQTLs from multiple tissue types.
"The software will allow people to do analyses that connect genetic variations to phenotypes. That's really the key challenge facing the entire field," He said. "We now have a much better tool to make those connections."
Additional authors on the study, "Adjusting for genetic confounders in transcriptome-wide association studies leads to reliable detection of causal genes," include Siming Zhao, Wesley Crouse, Sheng Qian, and Kaixuan Luo from the University of Chicago.
More information: Adjusting for genetic confounders in transcriptome-wide association studies improves discovery of risk genes of complex traits, Nature Genetics (2024). DOI: 10.1038/s41588-023-01648-9
Journal information: Nature Genetics
Provided by University of Chicago



Post comments