
Cells are the basic units of life, and their spatial distribution and composition within tissues determine the structure and function of a system. For example, in neural tissues, neurons and glial cells are arranged in a specific manner to ensure efficient transmission of information; in the liver, different types of cells (e.g., hepatocytes and Kupffer cells) form anatomically specific structures that perform both metabolic and immune functions. In the tumor microenvironment, the interactions and spatial remodeling of tumor cells, immune cells and stromal cells can influence tumor growth, invasion and metastasis. However, the tumor microenvironment is an extremely complex and heterogeneous system, with different cell populations exhibiting diverse spatial distributions and compositions at different stages of the tumor and under different microenvironmental conditions, and this heterogeneity greatly restricts the comprehensive resolution of its functions. Therefore, the identification of spatial regions with specific cellular distribution and composition, so-called “spatial domains”, is crucial for revealing the function of the tumor microenvironment. These spatial domains may become key cell signaling and interaction “hotspots”, and by studying these hotspots, researchers can identify key biomarkers and potential therapeutic targets that affect tumor growth and metastasis.
In recent years, the rise of spatially resolved transcriptomics technologies has greatly advanced our understanding of cellular composition and function in complex tissues such as tumor microenvironments. These techniques enable researchers to probe the gene expression profiles of cells at the single-cell level, revealing their heterogeneity and functional diversity at specific spatial locations. However, great challenges remain in identifying pathology-relevant spatial domains from spatial transcriptomic data. On the one hand, traditional clustering methods rely on spatial proximity and gene expression data to identify specific expression patterns in different regions, thus revealing their pathological functions, but often neglect the functional associations and interactions between different cell types. On the other hand, the inverse convolution method, although able to infer the proportion of cell types in a specific region, does not fully consider the spatial continuity of these cells and their complex network of interactions.
On September 27, 2024, Dijun Chen's group at Nanjing University published a research paper entitled “SpaTopic: A Statistical Learning Framework for Exploring Tumor Spatial Architecture from Spatially Resolved Transcriptomic Data”. SpaTopic is a statistical learning framework for exploring tumor spatial architecture from spatially resolved transcriptomic data, which combines clustering and inverse convolutional analysis of spatial transcriptomic data through topic modeling to classify the tumor microenvironment into spatial domains with consistent cellular composition, thereby enabling fine-grained functional annotation. SpaTopic can accurately identify a variety of spatial domains related to tumor function, including tertiary lymphoid structures and tumor boundaries. More importantly, SpaTopic's inferred spatial domain markers are genetically stable, have good migration ability, and can be used to predict spatial domains in new datasets. In addition, SpaTopic supports quantitative comparison of spatial domains and functional analysis across datasets, providing a powerful tool and a new perspective for functional analysis of the tumor microenvironment, and its inferred spatial domain markers have a wide range of potential applications.
Briefly, SpaTopic utilizes spatial transcriptomics data and single-cell transcriptome data as inputs for spatial domain prediction, annotation and comparison. The analysis process is roughly as follows: first, the cell type composition of each spatial point (spot) is inferred using an inverse convolution method, and these spatial points are initially clustered by an unsupervised clustering method. Next, the Kolmogorov-Smirnov (KS) test was used to determine the cell type specificity scores for each subcluster. Then, the cell type specificity matrix was decomposed by applying the theme model to obtain two probability distribution matrices: a “theme-cell type matrix”, which represents the distribution of different cell types in each theme; and a “subcluster-theme matrix”, which represents the distribution of different cell types in each subcluster in different themes. The other is the “subcluster-theme matrix”, which represents the probability distribution of each subcluster in different themes. On this basis, the clusters-topics matrix is binarized and each cluster is assigned to a specific topic (called CellTopics). Through these steps, it is possible to map spatial point clusters to corresponding themes, where clusters belonging to a particular theme are defined as spatial domains. As a result, SpaTopic not only accurately characterizes cell types in spatial domains, but also enables quantitative comparisons between different spatial transcriptome datasets and mining of spatial gene expression patterns.
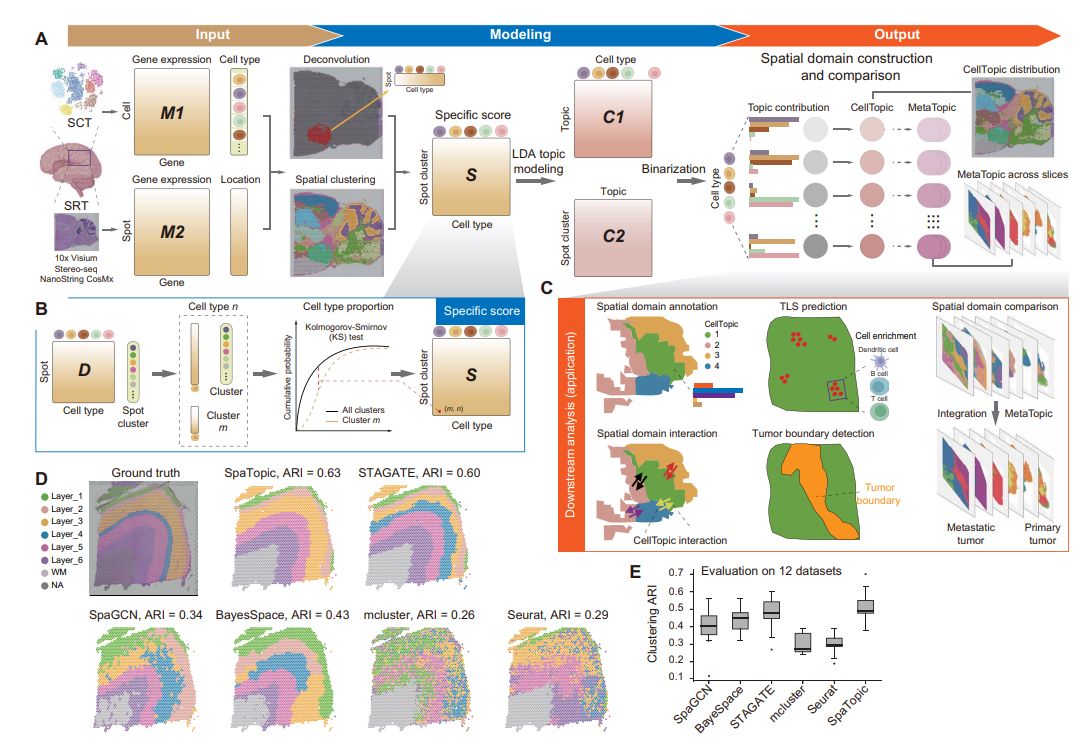
Figure 1. SpaTopic Technology Flowchart
The tertiary lymphoid structure is a unique immune microenvironment formed in non-lymphoid tissues as a result of chronic inflammation, and contains a wide range of immune cells such as B cells, T cells, and dendritic cells (DCs).The cellular composition of TLSs varies depending on the tissue and inflammatory conditions, posing a challenge for computational prediction. By analyzing spatial transcriptome data from primary liver cancer, SpaTopic identifies spatial domains associated with TLSs that are tightly co-localized with immune cells such as B cells, T cells, and dendritic cells, forming a typical TLSs structure. In addition, SpaTopic not only provides an unbiased approach for the identification of TLSs, but its derived marker gene set, TLS-25, shows a consistent expression pattern across cancer types, which can effectively predict the presence of TLSs and correlate with patient survival.
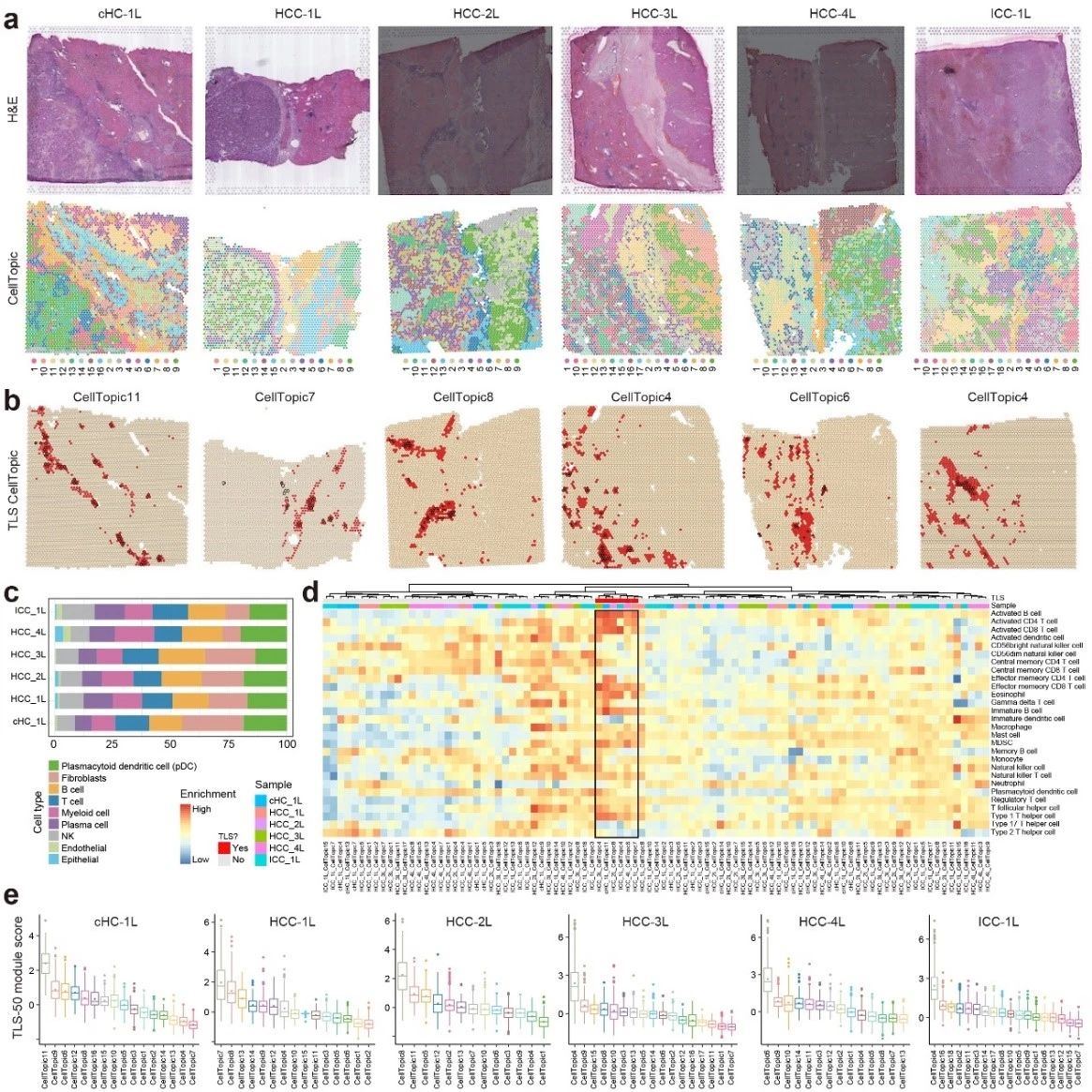
Figure 2. SpaTopic's accurate identification of TLSs
Further, SpaTopic's comparative analysis of the spatial cellular organization of colorectal cancer (CRC) primary tumors and liver metastases successfully identified different spatial domains of primary and metastatic tumors and grouped them into seven major MetaTopics through cluster analysis. the MetaTopics of primary and metastatic tumors exhibited consistent cellular composition, revealing the functional combinations of specific cell types. For example, the Mac_SPP1 cell subpopulation from MetaTopic2 (M2) was highly enriched in primary tumors, while the Mac_CXCL9 cell subpopulation was increased in metastatic tumors M4 and M6. Shared and unique MetaTopics were found between primary and metastatic tumors, while specific MetaTopics (e.g., M6 and M7) were exclusive to metastatic tumors. These findings are consistent with the results of single-cell data analysis and provide further evidence of cellular compositional and functional differences between primary and metastatic tumors.
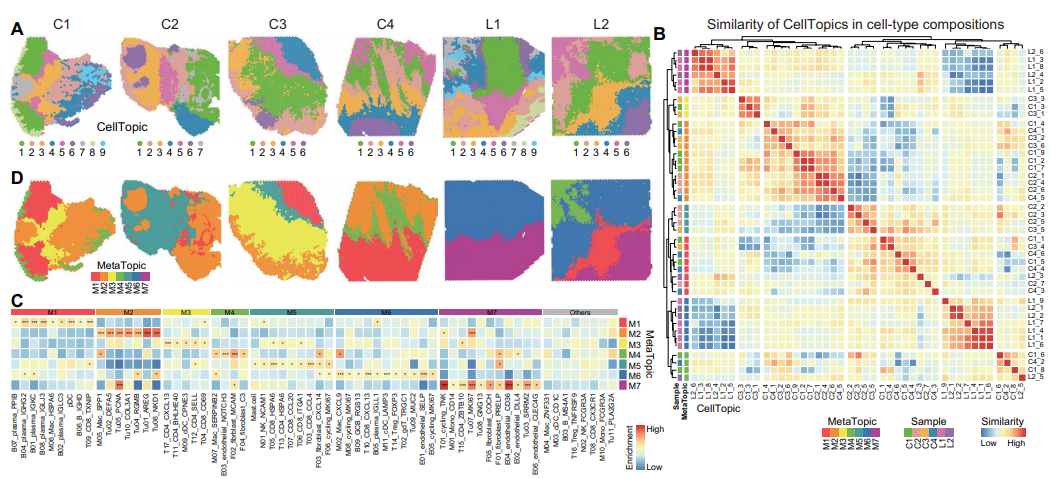
Figure 3. quantitative comparison of SpaTopic in the spatial domain of liver metastatic tumors in colorectal cancer
Then, through in-depth analysis of gene expression patterns among different MetaTopics, 907 genes were obtained that showed significant differences across MetaTopics, and they were classified into seven different gene modules (K1 to K7). For example, gene module K3 was significantly highly expressed in metastasis-specific MetaTopic M7, encompassing biological processes such as fatty acid metabolism and acute inflammatory response, which mark the characteristics of tumor metastasis. Modules K4 and K5 are active in primary tumor-related MetaTopics and are involved in functions such as energy production and metabolic regulation. Module K7 is shared between primary and metastasis-related MetaTopics and is enriched for immune-related pathways. By integrating six spatial transcriptome datasets, MetaTopics mapped explicitly in point-specific clustering of similar gene expression patterns, further validating the effectiveness of SpaTopic in quantitative comparison of spatial domains and functional annotation.

Figure 4. MetaTopics expression partitioning
Taken together, this study demonstrates the power of SpaTopic in parsing and annotating cellular spatial domains in the tumor microenvironment. By identifying spatial domains with consistent gene expression patterns and cell type composition, SpaTopic is not only able to reveal potential functional units in the tumor microenvironment, but also supports quantitative comparisons across samples and datasets, with good applicability.






Post comments