
by Greta Friar, Whitehead Institute for Biomedical Research

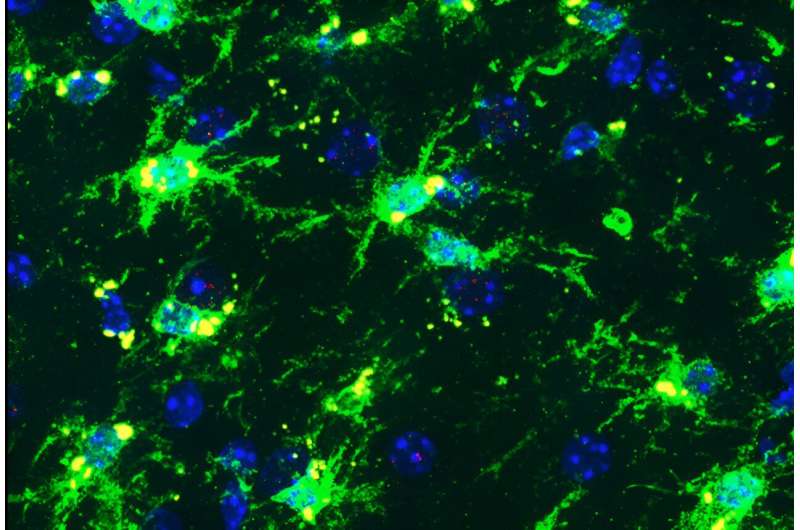
Smaller, more circularized mitochondria in glia of RTT cerebral organoids. Credit: Scientific Reports (2024). DOI: 10.1038/s41598-024-71040-y
Rett syndrome is a X-chromosome-linked neurodevelopmental disorder; it can lead to loss of coordination, mobility, ability to speak, and use of the hands, among other symptoms. The syndrome is typically caused by mutations within the gene MECP2.
Researchers in Whitehead Institute Founding Member Rudolf Jaenisch's lab have studied Rett syndrome for many years in order to understand the biological mechanisms that cause disease symptoms, and to identify possible avenues for treatments or a cure.
Jaenisch and colleagues have gained many insights into the biology of Rett syndrome and developed tools that can rescue neurons from Rett syndrome symptoms in lab models. However, much about the biology of Rett syndrome remains unknown.
New research from Jaenisch and postdoc Danielle Tomasello focuses on an understudied question: how Rett syndrome affects cell types in the human brain other than neurons. Specifically, Tomasello investigated the effects of Rett syndrome on astrocytes, a type of brain cell that supports and provides energy for neurons.
The work, published in Scientific Reports, details changes that occur in Rett syndrome astrocytes, in particular in relation to their mitochondria, and shows how these changes directly impact neurons. The findings provide a new framework for thinking about Rett syndrome and possible new avenues for therapies.
"By considering Rett syndrome from a different perspective, this project expands our understanding of a multifaceted and thus far incurable disease," says Jaenisch, who is also a professor of biology at the Massachusetts Institute of Technology.
Energy metabolism in Rett syndrome
Mitochondria are organelles that generate energy, which cells use to carry out their functions, and mitochondrial dysfunction was known to occur in Rett syndrome. Jaenisch and Tomasello found that mitochondria in astrocytes are particularly affected, even more so than mitochondria in neurons.
Tomasello grew human stem-cell-derived astrocytes in 2D cultures and also grew 3D organoids: mini brain-like tissues that contain multiple cell types growing in a structure that resembles actual brain anatomy. This approach allowed Tomasello to use human cells, rather than an animal model, and to study how cells behave within a brain-like environment.
When the researchers observed Rett astrocytes grown in these conditions, they found that the mitochondria were misshapen. They were short, small circles instead of large, long ovals. Additional studies showed evidence of the mitochondria experiencing stress and not being able to generate enough energy through their usual processes.
Because the mitochondria did not have enough of the typical proteins they use to make energy, they began to break down the cell's supply of the building blocks of proteins, amino acids, for parts to make up for the missing material. Additionally, the researchers observed an increase in reactive oxygen species, byproducts of mitochondrial metabolism that are toxic to the cell.
Further experiments suggested that the cells try to compensate for this mitochondrial stress by increasing transcription of mitochondrial genes. For example, Tomasello found that regions of DNA called promoters that can increase expression of key mitochondrial genes were more open for the cell to use in Rett astrocytes. Together, these findings paint a picture of severe mitochondrial dysfunction in Rett astrocytes.
Although mitochondria in Rett neurons did not have such severe defects, astrocytes and neurons have a close relationship. Not only do neurons rely on astrocytes to supply them with energy, they even accept mitochondria from astrocytes to use for themselves.
Jaenisch and Tomasello found that neurons take up dysfunctional mitochondria from Rett astrocytes at a higher rate than they take up mitochondria from unaffected astrocytes. This means that the effects of Rett syndrome on astrocytes have a direct effect on neurons: the dysfunctional mitochondria from the astrocytes end up in the neurons, where they cause damage.
Tomasello took mitochondria from Rett astrocytes and placed them on both healthy and Rett neurons. In either case, the neurons took up the dysfunctional mitochondria in large numbers and then experienced significant problems.
The neurons entered a hyperexcitable state that is ultimately toxic to the brain. The neurons also contained higher levels of reactive oxygen species, the toxic byproducts of mitochondrial metabolism, which can cause widespread damage. These effects occurred even in otherwise healthy neurons that did not themselves contain a Rett-causing MECP2 mutation.
"This shows that in order to understand Rett syndrome, we need to look beyond what's happening in neurons to other cell types," Tomasello says.
Learning about the role that astrocytes play in Rett syndrome could provide new avenues for therapies. The researchers found that supplying affected astrocytes with healthy mitochondria helped them to recover normal mitochondrial function.
This suggests to Tomasello that one possibility for future Rett syndrome therapies could be something that either targets mitochondria, or supplies additional mitochondria through the bloodstream.
Together, these insights and their possible medical implications demonstrate the importance of taking a broader look at the foundational biology underlying a disease.
More information: Danielle L. Tomasello et al, Mitochondrial dysfunction and increased reactive oxygen species production in MECP2 mutant astrocytes and their impact on neurons, Scientific Reports (2024). DOI: 10.1038/s41598-024-71040-y
Journal information: Scientific Reports
Provided by Whitehead Institute for Biomedical Research






Post comments