
by St. Jude Children's Research Hospital
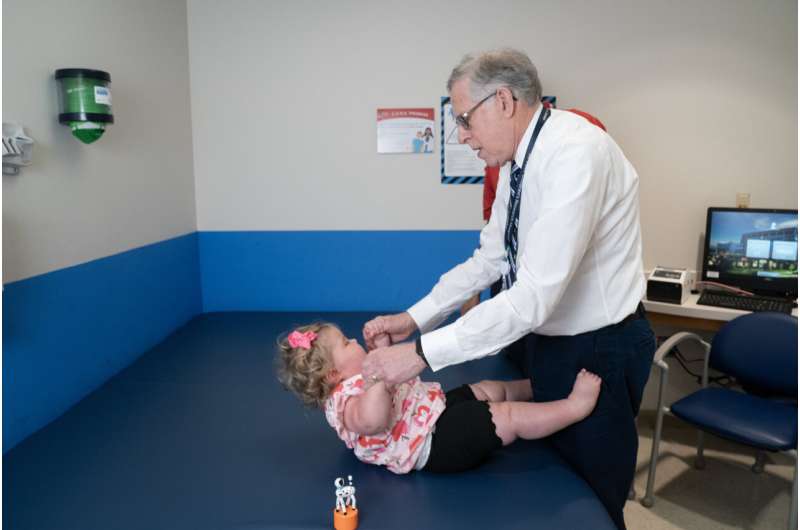
Prenatal therapy for spinal muscular atrophy (SMA) with risdiplam has shown promise in a first-of-its-kind study led by Richard Finkel, MD, St. Jude Center for Experimental Neurotherapeutics director and Department of Pediatric Medicine member, pictured. Credit: St. Jude Children's Research Hospital
Spinal muscular atrophy (SMA) is a progressive neurodegenerative disorder set in motion before birth. Scientists at St. Jude Children's Research Hospital led the first in utero treatment of SMA with the orally administered drug risdiplam.
More than two years after the child was born, no identifiable features of SMA have been observed. This study demonstrates the feasibility of treating SMA prenatally and supports further investigation into the approach. The findings were published in the New England Journal of Medicine.
"Our primary objectives were feasibility, safety and tolerability, so we're very pleased to see that the parent and child are doing well, " said the study's corresponding author Richard Finkel, MD, St. Jude Center for Experimental Neurotherapeutics director and Department of Pediatric Medicine member.
"The results suggest it would be worthwhile to continue investigating the use of prenatal intervention for SMA."
SMA is caused by a lack of survival motor neuron protein and occurs in around one in every 11, 000 births in the United States. If not treated, SMA type 1 (SMA-1), the most common and severe form, results in progressive muscle weakness that leads to death.
Currently, treatments for SMA-1 have demonstrated improved survival and motor function in infants, especially if administered shortly after birth before symptoms begin, but it is not a cure.
N-of-1: A case study of prenatal risdiplam
Survival motor neuron protein is most needed in the third trimester of fetal development and the first three months of life after birth. Symptom severity is, therefore, closely linked with the intervention time point.
Due to this clinical need, St. Jude researchers, as part of the Pediatric Translational Neuroscience Initiative, launched a unique clinical protocol to study risdiplam in a single patient. The goal was to determine the feasibility of starting treatment for a fetus with SMA-1 in utero.
The parents in this case were both known carriers of SMA genetic variants and had a prior infant born with SMA-1 before current treatments became available, who died at 16 months of age.
Genetic testing conducted by amniocentesis confirmed the fetus had no copies of the survival motor neuron gene, which, in combination with the family history and other genetic information, was highly predictive of the infant being born with SMA-1. Risdiplam was administered to the expectant mother during the final six weeks of pregnancy.
Safe and promising outcome encourages future research
Shortly after birth, the infant was diagnosed with three developmental abnormalities: ventricular septal defect (which resolved), optic nerve hypoplasia and a brainstem asymmetry, with related delays in vision and overall development. These abnormalities are considered to have occurred early in fetal development before exposure to risdiplam.
Now two-and-a-half years old, the child continues to be monitored periodically at St. Jude. "During the course of the assessment, we really have seen no indication of any signs of SMA, " Finkel said. The research demonstrates the safety and feasibility of the approach and bolsters the case for a more comprehensive study.
More information: Richard S. Finkel et al, Risdiplam for Prenatal Therapy of Spinal Muscular Atrophy, New England Journal of Medicine (2025). DOI: 10.1056/NEJMc2300802 Journal information: New England Journal of Medicine






Post comments