
The field of mitochondrial medicine faces significant challenges in evaluating the extent of brain disease in vivo, hindering our understanding of disease mechanisms and the identification of new biomarkers and therapeutic targets. Current imaging modalities like magnetic resonance spectroscopy (MRS) primarily focus on structural changes and biochemical markers, while little is known about the underlying metabolic mechanisms driving these changes. Pyruvate dehydrogenase deficiency (PDHD) serves as a case study, demonstrating the limitations of current imaging approaches. PDHD is a severe mitochondrial disease with a broad clinical spectrum, ranging from moderate neurological deficits to severe brain malformations. Patients typically present with elevated pyruvate and lactate levels, and while MRS can detect elevated lactate and decreased choline and N-acetyl-aspartate (NAA), it lacks the resolution to identify alternative substrates like acetate. Furthermore, the absence of standard imaging protocols to assess brain disease extent and the inconsistent use of MRS at diagnosis or during acute neurological deficits further complicate disease characterization. Consequently, the dynamic changes in the in vivo metabolic profile of the PDHD brain and the mechanisms driving these changes remain elusive.
The study by Marin-Valencia et al. explored the intricate metabolic network of the brain in PDHD. Utilizing a comprehensive approach that combined multi-modal imaging, isotope tracing, and metabolic profiling, the authors uncovered a distinct metabolic landscape in the PDHD brain, revealing potential therapeutic targets and pathways for intervention.
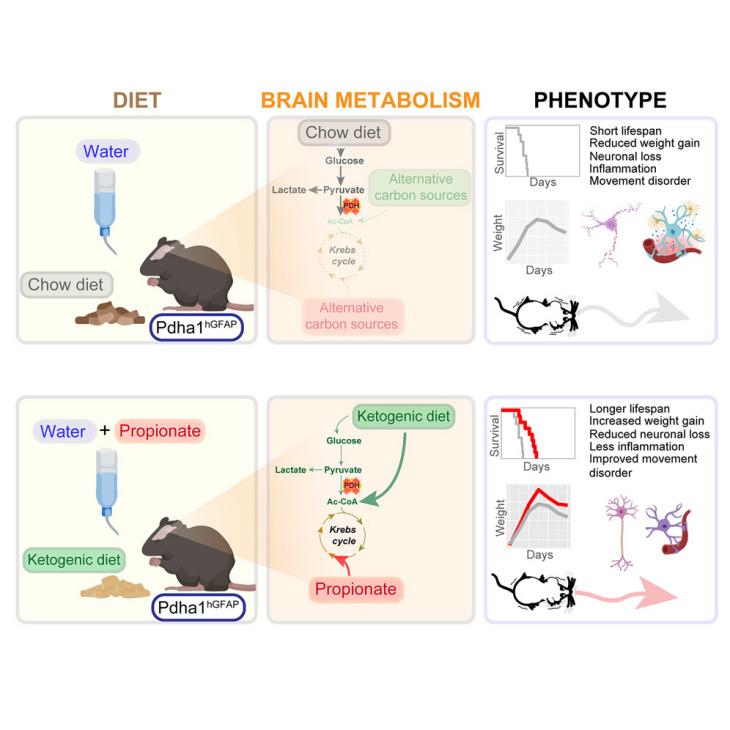
The study began by characterizing the neuropathological features of the Pdha1hGFAP mice, a PDHD mouse model, which closely resembled those observed in human patients, including microcephaly, cortical atrophy, and neuronal loss. Intriguingly, the authors observed a significant increase in brain glucose uptake and glycolysis as the disease progresses. This heightened glucose utilization was supported by the presence of reactive astrocytes, which expressed high levels of GLUT1, a key glucose transporter. The increased expression of GLUT1, along with blood vessel dilation, suggested a compensatory mechanism to supply glucose to the brain and meet the energy demands of damaged neurons.
Despite the increased glucose uptake, the authors found that glucose oxidation was impaired at the PDH level, a critical enzyme in the pyruvate dehydrogenase complex (PDC). This deficiency led to the accumulation of lactate and a decrease in the bicarbonate:lactate ratio, indicating a shift towards alternative metabolic pathways. The authors previously identified acetate as a potential alternative substrate to glucose, and this study uncovered another entry point: succinate-CoA. Succinate-CoA is primarily generated from propionate, a 3-carbon fatty acid produced by the gut microbiota and absorbed through monocarboxylate transporters.
In addition, the study employed 13C isotope tracing to track the metabolism of propionate in the PDHD brain. The 13C-labeling analysis revealed a significant increase in the incorporation of propionate into the brain, particularly in the cortex, hippocampus, and cerebellum of the Pdha1hGFAP mice. This suggested that propionate was being actively taken up and metabolized in the brain. The 13C-NMR isotopomer analysis revealed a distinct labeling pattern in the brain, with propionate primarily metabolized into succinate-CoA in glial cells. This was supported by the increased expression of propionate metabolism-related genes (Pcca, Pccb, and Mmut) in glial cells compared to neurons. The 13C-labeling pattern of aspartate showed a higher enrichment of the D23 carbon position, which was primarily derived from propionate metabolism. This suggested that propionate was contributing to the net synthesis of aspartate in the brain, a key metabolite involved in neurotransmission and brain development. Moreover, the labeling pattern of glutamate and glutamine, two key metabolites in the Krebs cycle, further supported the notion that propionate metabolism primarily occurred in glial cells, particularly those with PDH deficiency.
Subsequently, the study investigated the potential therapeutic benefits of propionate supplementation in combination with a ketogenic diet (KD). The results were promising: Pdha1hGFAP mice fed a KD and supplemented with propionate exhibited extended lifespan, reduced neuronal loss, and improved motor function compared to those on a standard chow diet. This suggested that propionate, in conjunction with a KD, can effectively compensate for the metabolic defects in PDHD and potentially alleviate the disease's severity.
In summary, the study provides valuable insights into the metabolic reprogramming of the PDHD brain, highlighting the pivotal role of glucose uptake, glycolysis, and alternative substrates like propionate in maintaining brain energy metabolism. The findings also open doors for exploring the mechanisms by which propionate modulates inflammation and supports neuronal survival in PDHD and other neuroinflammatory disorders. Future studies should focus on optimizing propionate dosing, determining potential side effects, and investigating the cellular and molecular mechanisms underlying propionate's therapeutic effects. Additionally, clinical trials are needed to assess the efficacy and safety of propionate-based treatments in PDHD patients.
Reference:
Marin-Valencia I, Kocabas A, Rodriguez-Navas C, Miloushev VZ, González-Rodríguez M, Lees H, Henry KE, Vaynshteyn J, Longo V, Deh K, Eskandari R, Mamakhanyan A, Berishaj M, Keshari KR. Imaging brain glucose metabolism in vivo reveals propionate as a major anaplerotic substrate in pyruvate dehydrogenase deficiency. Cell Metab. 2024 Jun 4;36(6):1394-1410.e12.






Post comments