
Credit:A mitochondrial NADPH-cholesterol axis regulates extracellular vesicle biogenesis to support hematopoietic stem cell fate.
NADPH, a vital coenzyme in cellular metabolism, plays a crucial role in driving reductive biosynthesis and antioxidant defense. Despite its essential functions, understanding the regulation of NADPH production and utilization within different cellular compartments remains a challenge. Recently, Niu et. al. employed a novel deuterium labeling strategy to investigate the compartmentalization of NADPH metabolism and its regulation within cells.
The authors leveraged the distinct localization of proline biosynthesis in the cytosol and mitochondria. In the cytosol, proline is synthesized from the reduction of pyrroline-5-carboxylate (P5C) using NADPH as a cofactor. In contrast, mitochondrial proline synthesis utilizes NADH as a cofactor. This differential cofactor usage allowed the authors to trace deuterium incorporation from labeled glucose into proline metabolites as a proxy for NADPH flux within each compartment.
The authors first demonstrated the feasibility of their approach using isocitrate dehydrogenase (IDH) mutants. IDH1 and IDH2, localized to the cytosol and mitochondria respectively, catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate, producing NADPH in the process. It was observed that mutations in IDH1 and IDH2 impaired NADPH production and instead led to the accumulation of the oncometabolite 2-hydroxyglutarate (2HG). By labeling cells with 3-2H glucose or 4-2H glucose, which label cytosolic and mitochondrial NADPH pools respectively, the authors observed that IDH1 mutations specifically affected NADPH flux in the cytosol, while IDH2 mutations impacted mitochondrial NADPH flux. These findings suggested that NADPH metabolism in the cytosol and mitochondria is independently regulated.
To further validate their approach and extend their findings to additional cell lines, the authors examined IDH1 and IDH2 mutant cells from GL261 murine glioma and TF-1 erythroleukemia cell lines. Consistent with their previous results, they found that IDH1 and IDH2 mutations in these cell lines also caused compartment-specific alterations in NADPH flux.
To investigate the impact of NADPH challenges on cellular metabolism, the authors introduced a water-generating NADPH oxidase (TPNOX) into the cytosol or mitochondria of HCT116 cells. TPNOX depletes NADPH levels in the compartment where it is expressed. They observed that cytosolic TPNOX expression altered cytosolic NADPH flux, while mitochondrial TPNOX expression affected mitochondrial NADPH flux, further supporting the compartmentalization of NADPH metabolism.
The authors also explored the use of chemotherapeutic drugs cisplatin and doxorubicin as tools to perturb NADPH metabolism. These drugs generate reactive oxygen species (ROS) in mitochondria, which require NADPH for neutralization. They found that low concentrations of cisplatin and doxorubicin selectively increased mitochondrial ROS levels without affecting cytosolic ROS. This led to alterations in mitochondrial NADPH flux, but not cytosolic NADPH flux, demonstrating the compartment-specific impact of these drugs on NADPH metabolism.
This study provides compelling evidence for the independent regulation of NADPH metabolism in the cytosol and mitochondria. The authors' deuterium labeling approach offers a powerful tool for investigating compartmentalized metabolism and reveals that cells respond to NADPH demands by specifically increasing the flux of NADPH-producing pathways within the same compartment. This lack of intercompartmental NADPH transfer suggests that cells have limited metabolic flexibility in supporting NADPH demands, which may create compartmentalized redox vulnerabilities exploitable for therapeutic purposes.
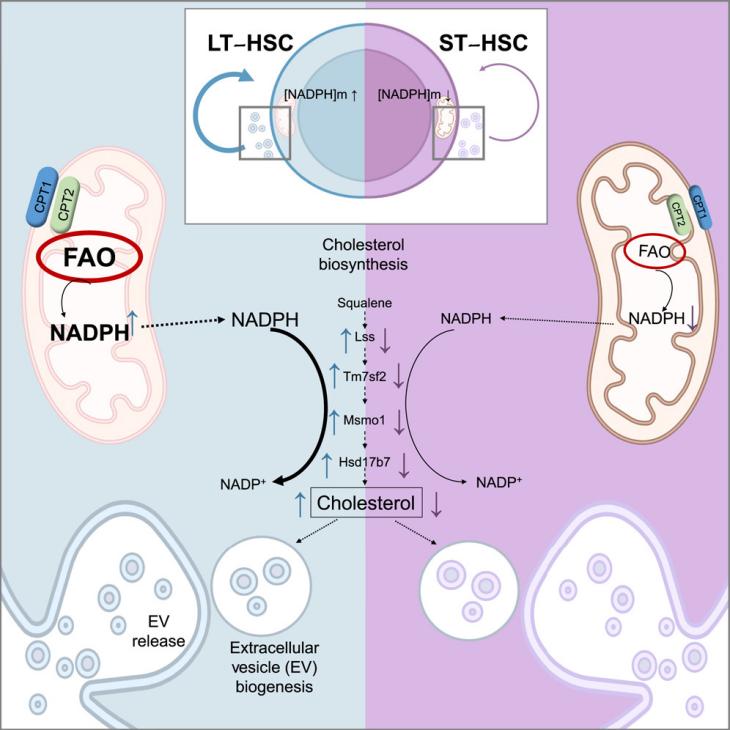
Independent regulation of NADPH metabolism confers specific functions on NADPH in different compartments, as recently discovered in a study conducted by Bonora et. al.. They uncovered a previously unrecognized axis involving mitochondrial NADPH, cholesterol, and extracellular vesicles (EVs) that played a crucial role in regulating HSC self-renewal and hematopoietic homeostasis.
The study began by investigating the mitochondrial NADPH levels in HSCs. Using flow cytometry and imaging techniques, the authors found that primitive HSCs, particularly those lacking CD34 (CD34-) and expressing EPCR, possessed significantly higher mitochondrial NADPH levels compared to more differentiated cells. This observation was supported by biochemical assays and genetic inactivation of fatty acid oxidation (FAO), which disrupted the inheritance of mitochondrial NADPH during HSC division, leading to impaired self-renewal and reconstitution capacity.
The authors then explored the functional significance of this high NADPH level in HSCs. Through bioinformatic analysis of HSC transcriptomes and metabolomic profiling, they identified cholesterol biosynthesis as a key pathway supported by NADPH. Further studies confirmed that HSCs exhibit high cholesterol synthesis, which is essential for their self-renewal. Inhibiting cholesterol synthesis using pharmacological inhibitors or genetic deletion of FAO components significantly impaired HSC function, highlighting the critical role of this pathway.
Building upon these findings, the authors hypothesized that mitochondrial NADPH-driven cholesterol synthesis may support the biogenesis of EVs, which are known to play a role in cell-to-cell communication and HSC maintenance. They observed that HSCs actively release EVs, which can be transferred between HSCs and enhance their self-renewal potential. Importantly, the authors found that the FAO-NADPH-cholesterol axis is required for the proper loading of tetraspanins like CD9 and CD63 into HSC-derived EVs, suggesting a specific cargo selection mechanism.
To further explore the role of EVs, the authors used pharmacological inhibitors and genetic approaches to block EV formation. They found that inhibiting EV biogenesis impaired HSC self-renewal and reconstitution capacity, highlighting the importance of EVs in HSC function. Moreover, they demonstrated that HSC-derived EVs not only activated autocrine signaling within HSCs but also influenced the function of stromal cells within the bone marrow niche. Stromal cells preferentially uptake HSC-derived EVs and respond by increasing the expression of SCF, a cytokine essential for HSC maintenance.
In summary, this study reveals a novel mitochondrial NADPH-cholesterol axis that regulates EV biogenesis and release in HSCs. This axis is crucial for proper HSC self-renewal and hematopoietic homeostasis. The findings suggest that manipulating this axis could have therapeutic implications for HSC-related diseases and potentially improve HSC transplantation techniques. Further studies are needed to explore the specific mechanisms of EV cargo selection and the interplay between HSCs and other bone marrow cells in EV-mediated communication.
Reference:
Niu X, Stancliffe E, Gelman SJ, Wang L, Schwaiger-Haber M, Rowles JL 3rd, Shriver LP, Patti GJ. Cytosolic and mitochondrial NADPH fluxes are independently regulated. Nat Chem Biol. 2023 Jul;19(7):837-845.
Bonora M, Morganti C, van Gastel N, Ito K, Calura E, Zanolla I, Ferroni L, Zhang Y, Jung Y, Sales G, Martini P, Nakamura T, Lasorsa FM, Finkel T, Lin CP, Zavan B, Pinton P, Georgakoudi I, Romualdi C, Scadden DT, Ito K. A mitochondrial NADPH-cholesterol axis regulates extracellular vesicle biogenesis to support hematopoietic stem cell fate. Cell Stem Cell. 2024 Mar 7;31(3):359-377.e10.







Post comments