
by CU Anschutz Medical Campus
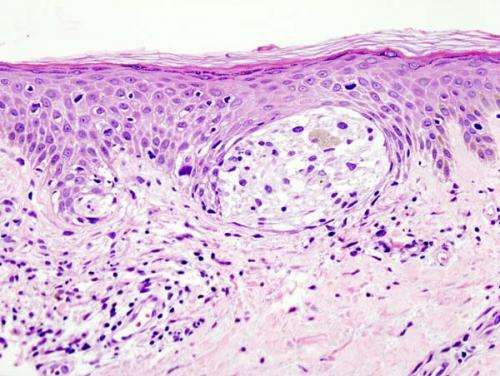
Melanoma in skin biopsy with H&E stain—this case may represent superficial spreading melanoma. Credit: Wikipedia/CC BY-SA 3.0
Think of the protein BH3 like a finger that turns off a cancer cell survival switch. The problem is that most cancer cells have found ways to remove this "finger—commonly, by breaking the action of a gene called p53 that puts the BH3 finger in motion. Now think of Bcl-2 as the switch itself. When cancer breaks p53, the BH3 finger never moves, and the Bcl-2 survival switch remains on. Despite thousands of published studies, researchers haven't had much luck directly protecting the action of p53. But, BH3 is another story. Drugs exist that mimic the action of BH3, collectively called (creatively...) "BH3 mimetics." For example, the drug venetoclax is a BH3 mimetic that has earned FDA approval against the blood cancer CLL and shows promise against the related blood cancer, ALL.
BH3 mimetics have also succeeded in the laboratory against the dangerous form of skin cancer, melanoma. But unlike blood cancers, it seems that melanoma has a sneaky way to work around BH3 mimetics.
Now, a University of Colorado Cancer Center, Department of Dermatology, study published in the journal Cell Death and Disease shows how melanoma escapes existing BH3 mimetics, and suggests a new strategy to block this avenue of escape.
The problem is that Bcl-2 itself is not the only switch that, when left in the "on" position, keeps cancer cells alive. BCL-2 describes a family of proteins, and even when Bcl-2 itself is turned off, melanoma cells can get equally effective survival signals from another member of the BCL-2 family, namely the protein MCL-1. The current study wondered what would happen if both these switches were turned off at the same time.
When the group used a cousin of venetoclax, called navitoclax, to silence Bcl-2 along with the investigational drug A-1210477 to silence MCL-1, melanoma cells died. Not only did the combination of navitoclax and A-1210477 kill melanoma cells and melanoma patient samples regardless of the specific mutations driving the disease or whether the patient had received previous treatments, but the combination also killed the melanoma-initiating cells (aka cancer stem cells) that often resist treatment to restart the disease.
"In cancer cells, there is a mix of pro-death and anti-death proteins. Depending on the balance, these cells live or die. By using navitoclax and A-1210477 to mute Bcl-2 and MCL-1, we remove anti-death proteins and, on balance, cancer cells die," says first author, Nabanita Mukherjee, Ph.D., faculty research associate in the CU School of Medicine Department of Dermatology. The work is published from the laboratories of Drs. David Norris and Yiqun Shellman.
In addition to Bcl-2 and MCL-1, there was a third genetic player in this story of the BCL-2 family "survival switch." In breast cancer, DRP-1 makes death proteins. But, the current study shows that in melanoma, DRP-1 has the opposite effect, making anti-death proteins. The combination of navitoclax and A-1210477 acted against DRP-1. In addition, when the group used CRISPR/Cas9 to completely mute DRP-1, melanoma cells were killed even more efficiently.
"This finding has not been reported before, and our results suggest that DRP-1 inhibition would cooperate with BH3 mimetics therapy in melanoma," Mukherjee says.
"Although the treatments for melanoma have advanced dramatically, alternative options are still needed, especially for the patients who do not respond to or relapse from current targeted or immunotherapies," says Mukherjee. "Our studies suggest that the concept of targeting multiple BCL-2 family members is worth exploring further for these patients."
More information: Nabanita Mukherjee et al, BH3 mimetics induce apoptosis independent of DRP-1 in melanoma, Cell Death & Disease (2018). DOI: 10.1038/s41419-018-0932-z
Journal information: Cell Death and Disease
Provided by CU Anschutz Medical Campus





Post comments