
by New York University
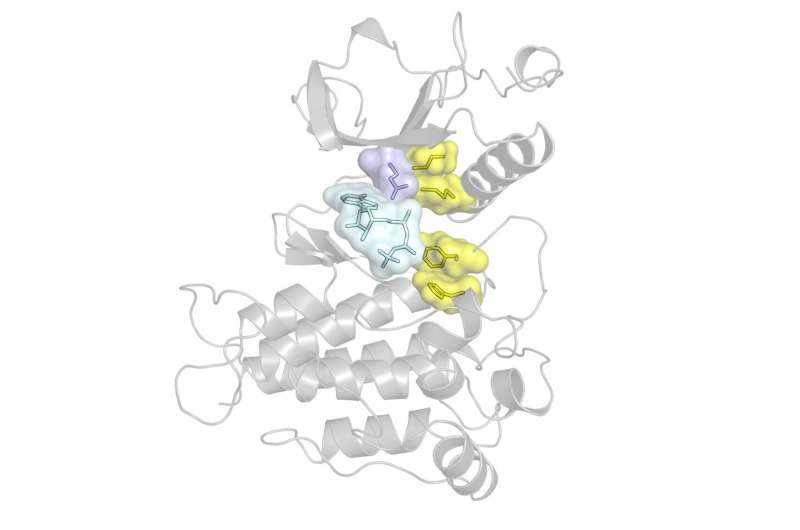
Structure of the FGFR kinase showing the locations of the gatekeeper residue in blue, hydrophobic spine in yellow, and ATP in cyan. Most drugs bind to the kinase in the same pocket as ATP. Credit: Traaseth Lab/NYU
More than 70 FDA-approved cancer drugs are kinase inhibitors, which work by blocking kinases—enzymes that add phosphate groups to molecules in the cell—and preventing the chemical activity necessary for signaling and growth in cancer cells. Kinase inhibitors can be very effective, but long-term, some patients experience aggressive recurrences that are more difficult to treat and resistant to the original drug.
A new study by New York University researchers, published this week in the journal Proceedings of the National Academy of Sciences, explains why cancers not only stop responding to kinase inhibitors but come back stronger, a finding that could inform which drugs oncologists use as a first-line treatment.
A leading cause of drug resistance—when a cancer no longer responds to a kinase inhibitor—is the emergence of genetic mutations, particularly those in a region in the kinase called the "gatekeeper" residue. The gatekeeper is deeply embedded in the kinase and allows or prevents access to an even-deeper hydrophobic (or water-repelling) pocket.
Because kinase inhibitors work by binding to this hydrophobic pocket, mutations to the gatekeeper residue block a drug's access, reducing its efficacy. But gatekeeper mutations also do something else that, according to the NYU researchers, may be even more important: they make kinases more active.
"When a kinase is switched into the active state, it can lead to processes like cells dividing, a hallmark of cancer," said Alida Besch, a Ph.D. student in NYU's Department of Chemistry and the study's first author. "This increased activity is why we hypothesize that cancers come back even stronger, but exactly how gatekeeper mutations increase kinase activity is not well understood."
In their study, the researchers focused on fibroblast growth factor receptors (FGFRs), a family of kinases that frequently mutates in different cancers, including lung and blood cancers. Treating FGFR-related cancers can involve using receptor tyrosine kinase inhibitors that bind to the hydrophobic pocket to block the receptor, which may effectively treat the cancer but also generate drug-resistant gatekeeper mutations.
Using a multipronged approach, including experiments (kinase activity assays and nuclear magnetic resonance or NMR spectroscopy) and computer simulations, the researchers studied FGFR kinases harboring two distinct gatekeeper mutations to determine how the kinase is made more active by the gatekeeper mutations.
Kinases need to convert from an inactive to active state in order to function, and prior research suggested that gatekeeper mutations affect the kinase's active state by strengthening and stabilizing a so-called "hydrophobic spine," a network of four residues connecting different areas in the kinase.
But the experiments and simulations revealed a different story: the researchers discovered that the gatekeeper mutations of the FGFR kinase actually affect the inactive state of the kinase, destabilizing it by weakening the hydrophobic spine, and therefore allowing the kinase to shift to the active form.
"This distinction—that gatekeeper mutations affect the kinase's inactive state and destabilize it—is important, because we generally want receptor tyrosine kinases to be held in the inactive state. Switching into the active state would usually be dictated by external signals like hormones, not the kinase itself," explained Yingkai Zhang, professor of chemistry at NYU and the Simons Center for Computational Physical Chemistry and the study's co-senior author.
"But if gatekeeper mutations are destabilizing the kinase and shifting it into its active form, this could explain why some cancers come back stronger."
The findings could potentially inform how clinicians choose which first-line cancer treatment to use—and whether a cocktail of drugs instead may be more effective at preventing recurrence.
"If a treatment targets kinases that we know will ultimately mutate, it might be better to use a cocktail treatment right away that will still bind to the kinase, even if the gatekeeper is mutated," said Nate Traaseth, professor of chemistry at NYU and the study's co-senior author.
The researchers are also considering how these findings could be used in the development of novel cancer therapeutics. One avenue they're exploring is finding locations in the kinase other than the hydrophobic pocket for drugs to bind to, given not only the prospect of gatekeeper mutations but also that these pockets look so similar in the roughly 500 different types of human kinases, which limits the chances that a drug can precisely target certain kinases.
"There's not a single kinase drug on the market that only hits one type of kinase, although that is the goal. Having drugs bind to different spots of the kinase that are more diverse than the hydrophobic pocket is one way to address this challenge," said Traaseth.
More information: Besch, Alida et al, Gatekeeper mutations activate FGF receptor tyrosine kinases by destabilizing the autoinhibited state, Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2213090120. doi.org/10.1073/pnas.2213090120
Journal information: Proceedings of the National Academy of Sciences
Provided by New York University






Post comments