
by University of Helsinki
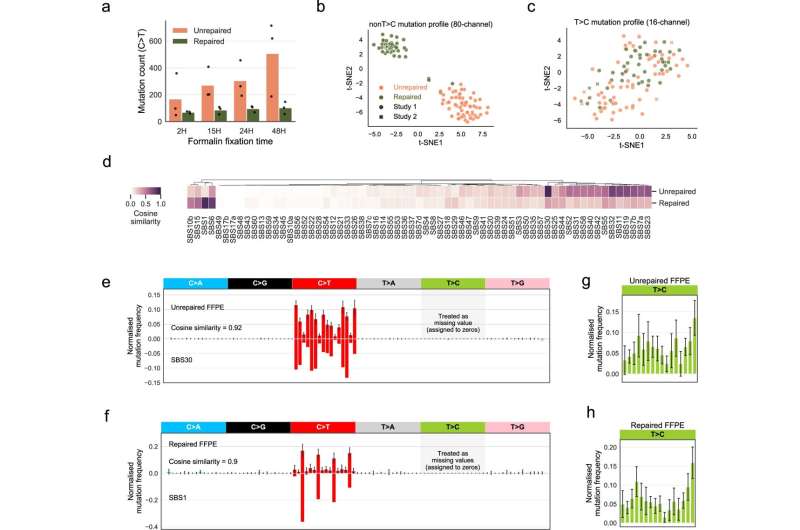
Mutational signatures of formalin exposure. a C>T FFPE-only mutation count increases with formalin fixation time. We observed this increase in both unrepaired and repaired-FFPE samples from study 1 (the fixation group). FFPE-only mutations refer to mutations that are only discovered in FFPE but not in FF samples or known germline databases. The bar height represents the average C>T count in n = 3 patients, and the individual counts are marked as black dots. b Consistent and separable mutational patterns observed for unrepaired-FFPE and for repaired-FFPE samples using 80-channel spectrum (non-T>C). We clustered the normalized 80-channel mutation profiles (n = 110) from study 1 and 2 using t-SNE (see Methods). c No consistent and separable mutational patterns observed for T>C mutations. We clustered the normalized T>C mutation profiles (n = 110) from study 1 and 2 using t-SNE. d Comparison of our derived FFPE signatures to known COSMIC SBS signatures. e, f Unrepaired signature is highly similar to SBS30 (e) and repaired signature is highly similar to SBS1 (f). We treated T>C features as missing data due to the strong batch-effect found in study 1, which is also observed in a few other studies shown in Supplementary Table 1 and therefore they were assigned to zeros. We noted that zero values are approximately close to the true T>C mutation probabilities in FFPE datasets without this batch-effect (Supplementary Fig. 6f). Error bars indicate the standard deviation in n = 55 independent samples with top 50% density in t-SNE cluster (see Methods). g, h Large variability in T>C mutation channels. We derived the T>C patterns using the same methods applied in (e, f). The error bar showed the standard deviations in n = 55 independent samples with top 50% density within the t-SNE (see Methods). Credit: Nature Communications (2022). DOI: 10.1038/s41467-022-32041-5
A new paper from University of Helsinki, published today in Nature Communications, suggests a method for accurately analyzing genomics data in cancer archival biopsies. This tool uses machine learning methods to correct damaged DNA and unveil the true mutation processes in tumor samples. This helps to unlock tremendous medicine values in millions of archival cancer samples.
Molecular-based diagnosis helps to match the right patient with the right cancer treatment. Researchers took particular interest in DNA profiling in clinical cancer samples.
"This invaluable source is currently not being used for molecular diagnosis due to the poor DNA quality. Formalin causes severe damage to DNAs, which therefore place an inevitable challenge to analyze cancer genomes in preserved tissues," says lead author Qingli Guo from University of Helsinki.
Analyzing mutation processes in cancer genomes can help early cancer detection, to accurately diagnose cancer, and reveal why some cancers become resistant to treatment. The new method can dramatically accelerate the development of clinical applications that can directly impact future cancer patient care.
The new method predicted more than 90% of developing cancer processes
Lead author Qingli Guo works in close collaboration with scientists from The Institute of Cancer Research (ICR), London, and Queen Mary University of London, developed machine learning methods, named FFPEsig, to unravel exactly how formalin mutates DNA.
"Our results show that normally nearly half of the cancer processes will be missed without noise correction. However, using FFPEsig, more than 90% of them were accurately predicted," says Qingli.
Cancer evolves gradually. Profiling mutational processes in longitudinal samples helps to identify clinical informative predictors and make diagnosis of each tumor stage.
"Our finding enables the characterization of clinically relevant signatures from the preserved tumors biopsies stored at room temperatures for decades. With a deep understanding of how formalin impacts cancer genome, our study opens a huge opportunity to transform the developed signature detection assays using the large cost-effective archival samples," say the researchers.
The researchers pointed out the method currently does not completely remove artifacts that appeared in FFPE samples showing batch effects, and how well the tool performs varies by cancer type, so care must be taken to interpret any findings. They are also interested in further applying their methods to a much broader spectrum of archival samples in the future.
More information: Qingli Guo et al, The mutational signatures of formalin fixation on the human genome, Nature Communications (2022). DOI: 10.1038/s41467-022-32041-5
Journal information: Nature Communications
Provided by University of Helsinki






Post comments