
by Hartwig Medical Foundation
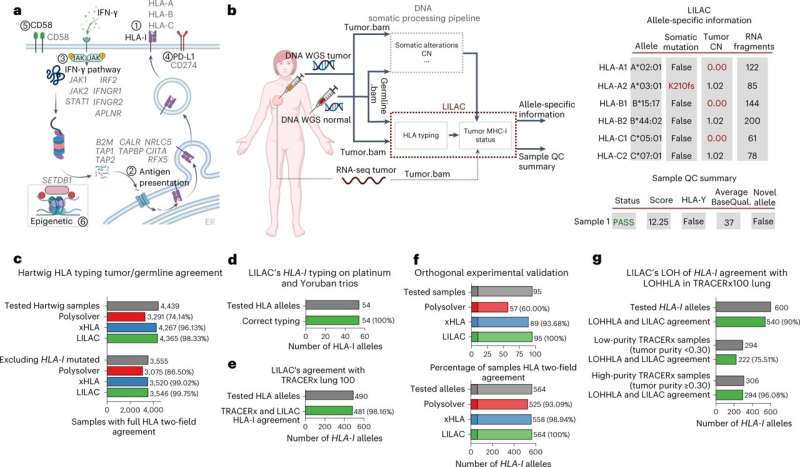
Inference of HLA-I tumor status with LILAC. a, Representation of the six immune escape pathways considered in the present study alongside their associated genes (adapted from ‘MHC class I and II pathways’, by BioRender.com). The genes considered for each immune escape pathway are depicted in gray. b, Left, workflow of the Hartwig tumor analytical pipeline integrating LILAC. LILAC’s framework is highlighted with red text and a red border. Right, tables showing an illustrative example of LILAC’s allele-specific and global patient reports (partially created with BioRender.com). QC, quality control; BaseQual., basecalling quality score. c, HLA-I typing tumor and germline agreement in Hartwig cohort. d, LILAC’s HLA-I typing validation using Platinum and Yoruban family trios. e, LILAC’s agreement with the HLA-I types from the TRACERx lung cohort. f, LILAC’s HLA-I typing experimental validation. g, LILAC and LOH of HLA-I agreement in the TRACERx100 lung cancer cohort. Credit: Nature Genetics (2023). DOI: 10.1038/s41588-023-01367-1
Cancer is caused by DNA changes that cause a cell to gradually change from benign to malignant. This can lead to metastases in other parts of the body. By analyzing the DNA data of more than 7,000 patients, the researchers show that there are major differences between primary and metastatic cancer and that there are also tumor types in which the primary tumor and the metastasis hardly differ from one another. By studying the types of DNA changes and the consequences of the changes, important insights into the underlying biological processes were obtained.
Researchers from UMC Utrecht, Vall d'Hebron in Barcelona and Hartwig Medical Foundation in Amsterdam and Australia have mapped the DNA changes of the 23 most common tumor types. They have studied the differences in genetic characteristics between the source of the cancer, the primary tumor, and metastatic tumors.
Unique collections of whole genome sequencing data from tumors were used. This enabled the researchers to study in great detail which changes in the tumor had occurred during and after the tumor had developed. The researchers have harmonized and systematically compared the world's largest publicly available data sets of primary tumors (from the international PCAWG consortium with information from ~2,800 patients) and metastatic tumors (Hartwig Medical Database, ~4,400 patients). The results of this research were published May 10 in the journals Nature and Nature Genetics.
The studies were initiated and coordinated by Prof. Dr. Edwin Cuppen (professor of human genetics at the Center for Molecular Medicine (CMM) and Oncode Institute at UMC Utrecht and scientific director of the Hartwig Medical Foundation in Amsterdam) and led by Dr. Francisco Martínez Jiménez, current head of VHIO's Computational Immunogenomics Group, and Arne van Hoeck, senior scientist at CMM, together with researchers from CMM and the Hartwig Medical Foundation in the Netherlands and Australia.
The paper published in Nature describes the overall genomic differences found when comparing primary and metastatic tumors and highlights the fact that the differences are highly dependent on the type of cancer studied, as well as the tumor's exposure to previous anti-tumor treatments.
One could state that this work confirms many observations that were previously done in cancer type-specific studies. However, the pan-cancer nature of the current study demonstrates which processes and mechanisms are shared between tumor types and also quantifies their prevalence per tumor type. Such a systematic analysis and comparison from a genome-wide perspective has never been performed before.
The second study, published in parallel in the journal Nature Genetics, presents an analysis of the genomic alterations that allow tumors to escape the immune system, as well as a comparison of their prevalence in primary and metastatic tumors.
The researchers found that the prevalence of genetic immune escape is highly variable between tumor types and that in certain tumor types only a single mechanism is present, while in others various processes were affected. Furthermore, they showed that there are not many differences between primary and metastatic tumors, indicating that immune evasion is a characteristic that is acquired relatively early in tumor development.
This is the first time a complete tumor genome-wide sequencing dataset has been generated for primary and metastatic tumors of this magnitude. These data are public and available for research, providing a new global resource for further research into the biology and evolution of cancer, as well as the development of new therapies to combat the disease.
Metastatic cancer genomics
Metastatic spread involves the detachment of tumor cells from a primary tumor, colonization of secondary tissue and growth in a hostile environment. Advanced metastatic tumors are often able to withstand aggressive treatment regimens and represent the leading cause of cancer-associated death. "Despite many efforts to understand these phenomena, we still have limited knowledge of the contribution of genomic changes to metastasis," says Martínez, first author of the two studies, which he developed at the Center for Molecular Medicine at the University of Utrecht.
It is therefore essential to characterize genomic differences between primary and metastatic cancers and quantify their impact on resistance to therapies in order to understand and exploit therapeutic interventions that establish more effective and personalized therapies.
To address these questions, researchers from the Center for Molecular Medicine and Oncode Institute at Utrecht University and Hartwig Medical Foundation have generated the largest harmonized complete genome sequencing dataset of tumor genomes from cancer patients. This dataset covers more than 7,000 unpaired primary and metastatic tumor samples from 71 cancer types, including 23 cancer types with a large representation in both clinical stages.
Identifying genomic clues to metastasis
"One of the major problems we have encountered is that in recent years tumors have been analyzed and sequenced with completely different analysis protocols, which makes an integrated analysis of the data very difficult. In this work we have processed the complete genome sequencing of more than 4,000 metastatic tumors, most of them previously treated, and reanalyzed 2,500 untreated, early-stage, independent primary tumor samples with the same protocol. In total we have pooled data from 7,108 tumor samples from patients with a wide range of cancer types," says Arne van Hoeck, senior researcher at the Center for Molecular Medicine at University Medical Center Utrecht.
The analysis of genomic differences between primary and metastatic tumors has led the researchers to several conclusions. "If I had to choose only one of the differences we have found, it would be that the differences are highly dependent on the type of tumor. In some types of tumors, such as pancreatic cancer, the genomic differences between primary and metastatic tumors are subtle. While in others, such as prostate, thyroid and some subtypes of breast cancer, there are very important genomic differences," says Martínez.
In addition, the exhaustive analysis has allowed the researchers to identify recurrent genomic patterns in metastatic tumors such as the presence of high genomic instability, greater enrichment of structural genomic alterations versus point mutations, and the presence of genomic alterations associated with the acquisition of resistance to treatment. However, hardly any driver alterations exclusively associated with the metastatic process could be identified.
"In this sense," explains Martínez, "our results confirm a dominant trend in the field of metastasis research, in which the process of metastasis cannot be explained by a specific genomic alteration, but rather by an evolutionary process in which the interaction of tumor cells with the microenvironment surrounding the tumor possibly plays a very relevant role."
However, this study has identified a set of novel genomic alterations that are enriched in metastatic tumors and that may be associated with the acquisition of resistance to certain cancer treatments. "It is an important first step, but dedicated clinical studies are needed to validate clinical relevance for patients," conclude the researchers.
Genomic alterations in the tumor that allow it to escape from the immune system
In parallel, researchers have studied how tumors are able to escape the surveillance imposed by the immune system at different stages of the disease. This ability to escape often involves tumor-specific genomic alterations in immune-related pathways. In the study published in the journal Nature Genetics, researchers used the cohort of more than 7,000 whole genome samples from tumors described above to identify the prevalence of genomic alterations associated with immune escape, as well as to determine whether there are differences between primary and metastatic tumors. "We know that tumors have the ability to be invisible to the immune system, but we wanted to understand which genomic alterations confer this ability and how frequently we detect them in different types of cancer and at different stages of tumor progression."
The results of this analysis revealed that one in four patients has genomic alterations in the tumor that are directly associated with escape from immune recognition. However, the researchers have again found substantial differences between different types of cancer, while in some, such as cervical carcinoma, more than half of the patients have these alterations; in others the prevalence is practically none.
On the other hand, the comparison between primary and metastatic tumors revealed that there are hardly any differences between the two stages, either in the frequency or in the type of immune system escape disorders observed. "This leads us to believe that most tumors probably acquire the ability to evade the immune system at very early stages of their evolution," explains Martínez.
Another feature found in this analysis is that the type and frequency of escape gene alterations is directly related to the number of mutations in the tumor (i.e., the number of changes in the DNA sequence of the tumor genome). In tumors with a low mutational load, virtually no escape gene alterations are observed, whereas in tumors with a medium and high mutational load, alterations that partially truncate recognition by the immune system are frequently observed. Finally, tumors with very high mutational load (commonly referred to as hypermutated or ultra-hypermutated) have a very specific type of alteration that tends to completely truncate recognition by the immune system.
"Now that we have a more complete picture of the landscape of immune escape gene alterations that make the tumor invisible to the immune system, the next step is to investigate whether these alterations have an influence on the immunotherapy treatment response," says Martínez.
Research leader Prof. Dr. Edwin Cuppen states, "The new insights from these studies form a valuable basis for improving diagnostics in cancer patients, personalizing therapeutic treatment and possibly preventing (metastatic) cancer. The results of the study also make clear how important the systematic collection and disclosure of data for reuse in the healthcare domain is. Without the cooperation and consent of patients, as well as national-scale collaboration with hospitals, these studies would have been impossible. And these novel valuable insights for the improvement of future oncological care would not have been possible."
More information: Francisco Martínez-Jiménez et al, Pan-cancer whole-genome comparison of primary and metastatic solid tumours, Nature (2023). DOI: 10.1038/s41586-023-06054-z
Francisco Martínez-Jiménez et al, Genetic immune escape landscape in primary and metastatic cancer, Nature Genetics (2023). DOI: 10.1038/s41588-023-01367-1
Journal information: Nature Genetics , Nature
Provided by Hartwig Medical Foundation






Post comments