
by University of Barcelona
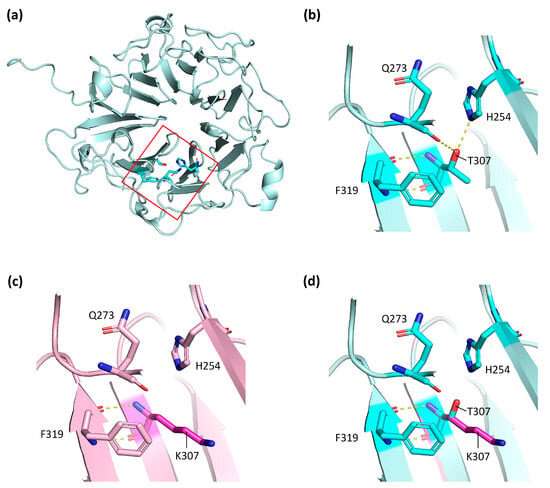
In silico studies of c.920C>A;p.(Thr307Lys) variant in the RPGR gene. Credit: Cells (2024). DOI: 10.3390/cells13060524
Ciliopathies are rare diseases in which the formation or function of cilia, cylindrical-shaped extensions found on the surface of many cells, is altered. There is a high degree of ciliary specialization, ranging from motile cilia of the respiratory epithelium to primary cilia—necessary for neurodevelopment or the formation of many organs—and neurosensory cilia of the ear and the retina. Some proteins are common to several types of cilia while others are tissue-specific, so ciliopathies may or may not be associated with a particular syndrome.
A respiratory ciliopathy that can commonly occur in combination with alterations in other tissues with motile cilia is primary ciliary dyskinesia (PCD). However, in some families, this pathology can also occur together with retinitis pigmentosa, an inherited vision disease.
Now, a paper published in the journal Cells has identified a potential modifier gene for PCD by studying a family in which the mother and two sons are affected by X-linked retinitis pigmentosa (XLRP), and one of the sons also has primary ciliary dyskinesia (PCD).
The work is coordinated by Professor Gemma Marfany, from the Faculty of Biology and the Institute of Biomedicine of the University of Barcelona (IBUB), and Núria Camats-Tarruella, from the Vall d'Hebron Research Institute (VHIR); both are members of the Center for Biomedical Research Network on Rare Diseases (CIBERER). The research, in which a team from the Andalusian Public Foundation for Progress and Health of Andalusia is collaborating, has been carried out under an intramural cooperative and complementary action (ACCI) of the CIBERER.
The genetic variant causing retinitis pigmentosa in the family
Mutations in the RPGR gene cause more than 70% of cases of XLRP. This gene has alternative splicing (cutting and splicing of genetic material) that is specific to the retina, but also produces a protein isoform—one of several forms of the same protein—expressed in all hair cells.
In the new study, the team studied whether a missense mutation in the RPGR gene—potentially responsible for retinitis pigmentosa in the two sons and the mother—could explain FAPD in only one of the men, or whether there might be other variants in modifier genes that explain the phenotypic variability in the family.
Immunofluorescence of nasal epithelial samples from siblings and the carrier mother showed that expression of the mutated allele correlated with decreased localization of the RPGR gene in the cilia transition zone and its delocalization in the cytoplasm. This was corroborated by in vitro expression assays.
The search for new variants in the person affected by SCD identified heterozygous missense variants in the CEP290 gene, which also causes syndromic ciliopathy. The localization of the CEP290 protein in the ciliary transition zone was also affected in patients, so the variant in the RPGR gene could be related to an alteration in the trafficking and localization of ciliary proteins.
These findings reveal a functional relationship between the two proteins, "which is why the CEP290 gene should be analyzed as a potential modifier of the respiratory phenotype in families with cases of SCD and XLRP," the team concludes.
More information: Noelia Baz-Redón et al, Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene, Cells (2024). DOI: 10.3390/cells13060524
Provided by University of Barcelona







Post comments