
by Tel-Aviv University
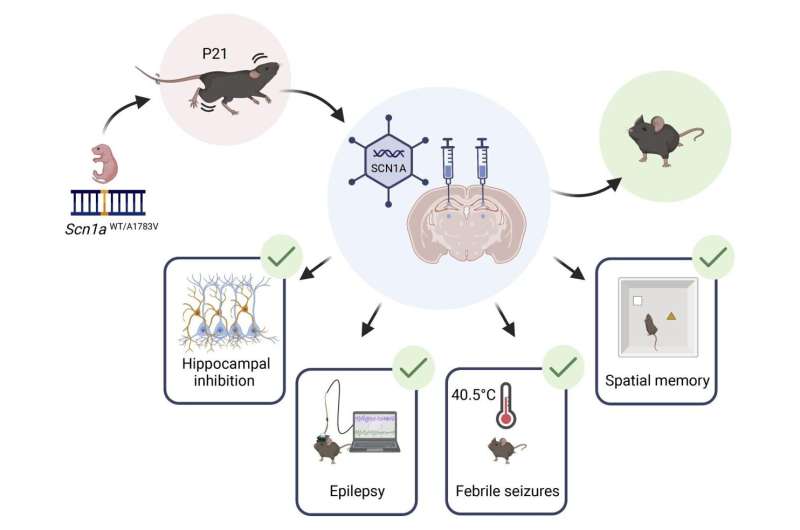
Graphical abstract. Credit: Journal of Clinical Investigation (2023). DOI: 10.1172/JCI159316
An innovative gene therapy may help treat a severe and fatal developmental epilepsy syndrome that affects children. Researchers hope that this genetic therapy can also be adapted to other types of genetic epilepsies and think that the tools developed in this research will pave the way for the development of similar treatments for other rare diseases.
Researchers at Tel Aviv University, among other institutions, have developed an innovative gene therapy that may help children suffering from Dravet syndrome (DS)—a severe developmental epilepsy that results from a mutation not inherited from the parents, but occurs randomly in the fetus, in a gene called SCN1A. Dravet syndrome manifests as severe epilepsy that is not well controlled by medication, together with developmental delays, cognitive impairment, and a high chance of early death.
As part of the study, a virus carrying a normal SCN1A gene was injected into the brains of DS mice, and the treatment was found to be effective in a variety of critical aspects: improvement in epilepsy, protection from early death, and significant improvement of cognitive abilities. Importantly, this treatment was effective after the onset of severe epilepsy in Dravet.
According to the researchers, "We hope that the technique we developed in the laboratory will also reach the clinic in the future and help children with this serious disease. In addition, since there is a similarity between Dravet and other rare developmental epilepsies, in terms of the patient's symptoms and brain changes, we hope that this treatment can also help other types of genetic epilepsies, and we think that the tools we developed in this research will pave the way for the development of similar treatments for other rare diseases."
The research was conducted under the leadership of Dr. Moran Rubinstein and graduate student Saja Fadila, along with Anat Mavashov, Marina Brusel and Karen Anderson, all from the Sackler Faculty of Medicine and the Sagol School of Neuroscience at Tel Aviv University, and Dr. Eric Kremer, from the University of Montpellier in France.
Also participating in the study were Bertrand Beucher and Iria González-Dopeso Reyes from Montpellier and other researchers from France, the U.S. and Spain. The research was published in the Journal of Clinical Investigation.
According to Dr. Rubinstein, "Dravet syndrome, whose incidence is approximately one in 16,000 births, is considered relatively common among rare genetic diseases. As of today, there are approximately seventy children living in Israel who suffer from DS. It is a severe developmental epilepsy, which begins with thermally-induced seizures around six months of age, and progresses, after one year of age, to frequent spontaneous epileptic seizures along with motor and cognitive developmental delays."
"The existing drugs for epilepsy do not sufficiently help children with DS, who are under significant risk of early death. It is known that this severe syndrome results from a genetic mutation that is not inherited from the parents, but is created randomly in the fetus in a gene called SCN1A. In addition, the disease is not characteristic of a certain segment of the population, cannot be predicted in advance, nor can it be discovered during pregnancy."
The researchers add, "It is customary nowadays to perform a genetic analysis for children who suffer from complex thermally-induced seizures around the age of six months. However, even if the test detects that the problem is in the SCN1A gene, the final diagnosis is often given after the epilepsy worsens, with the appearance of severe spontaneous convulsions and developmental delays."
"Although it is important to have an early diagnosis, diagnosis is often delayed, and most children are diagnosed only at the age of one or two years and sometimes even later."
"Recently developed genetic therapies have been found to be effective in DS mice, and some of them are even now in the phase of clinical trials in humans. However, these treatments have demonstrated efficacy in DS mice only when given at very early stages, even before the onset of symptoms. Since gene therapy is a complex and invasive procedure, it will not be given to children without a certain diagnosis of DS."
"Therefore, in this study, we concentrated on developing a treatment that would be effective after the onset of seizures, even at a relatively late age. In addition, since the syndrome also includes developmental delays, we sought to develop a treatment that would alleviate both the epilepsy and the cognitive symptoms."
Dr. Rubinstein adds, "In genetic therapies, it is customary to use viruses as carriers that carry normal genetic material into the patient's body in order for it to be added to the damaged DNA and enable normal activity. For this purpose, the virus is engineered: its original genetic material is removed so it cannot cause disease or replicate itself, and instead, the relevant normal gene is packed inside."
"In the case of Dravet syndrome, since the SCN1A gene is very large, it was not possible to use common viruses that are usually used for this purpose and a virus capable of carrying and transferring large genes was needed. In our study, we solved this problem by using a virus called Canine adeno virus type 2, as a carrier of the normal gene."
Next, the researchers injected the carrier virus directly into the brains of DS mice in order for the virus to infect the malfunctioning nerve cells. The researchers explain that the treatment requires direct injection into the brain because the size and properties of the virus do not allow it to pass through the blood-brain barrier.
Thirty one mice were treated at three weeks of age, after the onset of spontaneous convulsions (equivalent to one to two years of age in children), and thirteen mice were treated at five weeks of age (equivalent to approximately six to eight years of age in children). The injection was performed in several areas of the brain, and in addition an empty virus was injected into the brains of forty eight mice for control.
The results were promising: the highest efficacy was observed when the treatment was injected at three weeks of age. In these mice, the seizures stopped completely within just sixty hours of injection, life expectancy increased significantly, and the cognitive impairment (diagnosed using spatial memory tests) was fully repaired.
Even in mice treated at five weeks of age, a significant improvement was observed, which was manifested in a decrease in epileptic activity and protection from thermally-induced seizures. For the mice in the control group mice that received an empty virus, no improvement was observed and they suffered from the symptoms of the disease just like untreated mice, and about 50% of them died an early death as a result of the severe epilepsy. In addition, the treatment was applied to healthy mice with no harmful results—proof of its safety.
The researchers explain, "Our treatment added a normal gene to the damaged neurons in the brain, which was enough to restore them to normal function. The return of the normal gene in its entirety is particularly important for treating Dravet syndrome, because in different children the mutation occurs in different places in the gene, and injecting a complete gene is a uniform treatment suitable for all DS patients."
"In addition, we found that the virus chosen for the purpose of the study infects many nerve cells in the brain, and spreads widely beyond the injection site, adding to its effectiveness."
Dr. Rubinstein concludes, "The treatment we developed is the first that has been proven to be effective for Dravet syndrome when given after the onset of spontaneous convulsions, and the first that resulted in an improvement in the cognitive function of the DS mice. We registered a patent, and we hope that in the future the treatment will reach the clinic and help children suffering from this serious disease."
"In addition, we are currently investigating whether it may also be suitable for other genetic neurodevelopmental diseases. The platform we developed is a plug & play platform for genetic therapies, and perhaps in the future, we will be able to pack into the carrier virus different types of normal genetic material to treat additional diseases."
More information: Saja Fadila et al, Viral vector–mediated expression of NaV1.1, after seizure onset, reduces epilepsy in mice with Dravet syndrome, Journal of Clinical Investigation (2023). DOI: 10.1172/JCI159316
Journal information: Journal of Clinical Investigation
Provided by Tel-Aviv University







Post comments