
By:
Louis A. Cona, MD
Reviewed:
Robert J. Hancock
Delve into the 7 stages of ALS, from early symptoms to the final decline, and explore how stem cell therapy is emerging as a hopeful avenue for ALS treatment. This guide provides a detailed walkthrough of ALS progression, the impact on individuals, and the potential promise of stem cell interventions.

Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig's disease, is a neurodegenerative disorder characterized by the progressive degeneration of motor neurons.
The loss of motor neurons leads to the inability of the brain to initiate and control muscle movement, which can result in the loss of voluntary muscle action.
Key Takeaways:
Early detection of ALS can aid in better management of symptoms.
ALS progression is categorized into stages, each with unique symptoms and challenges.
Accurate diagnosis requires a mix of clinical examinations and tests.
Knowledge of ALS stages helps patients, families, and healthcare providers prepare for the journey ahead.
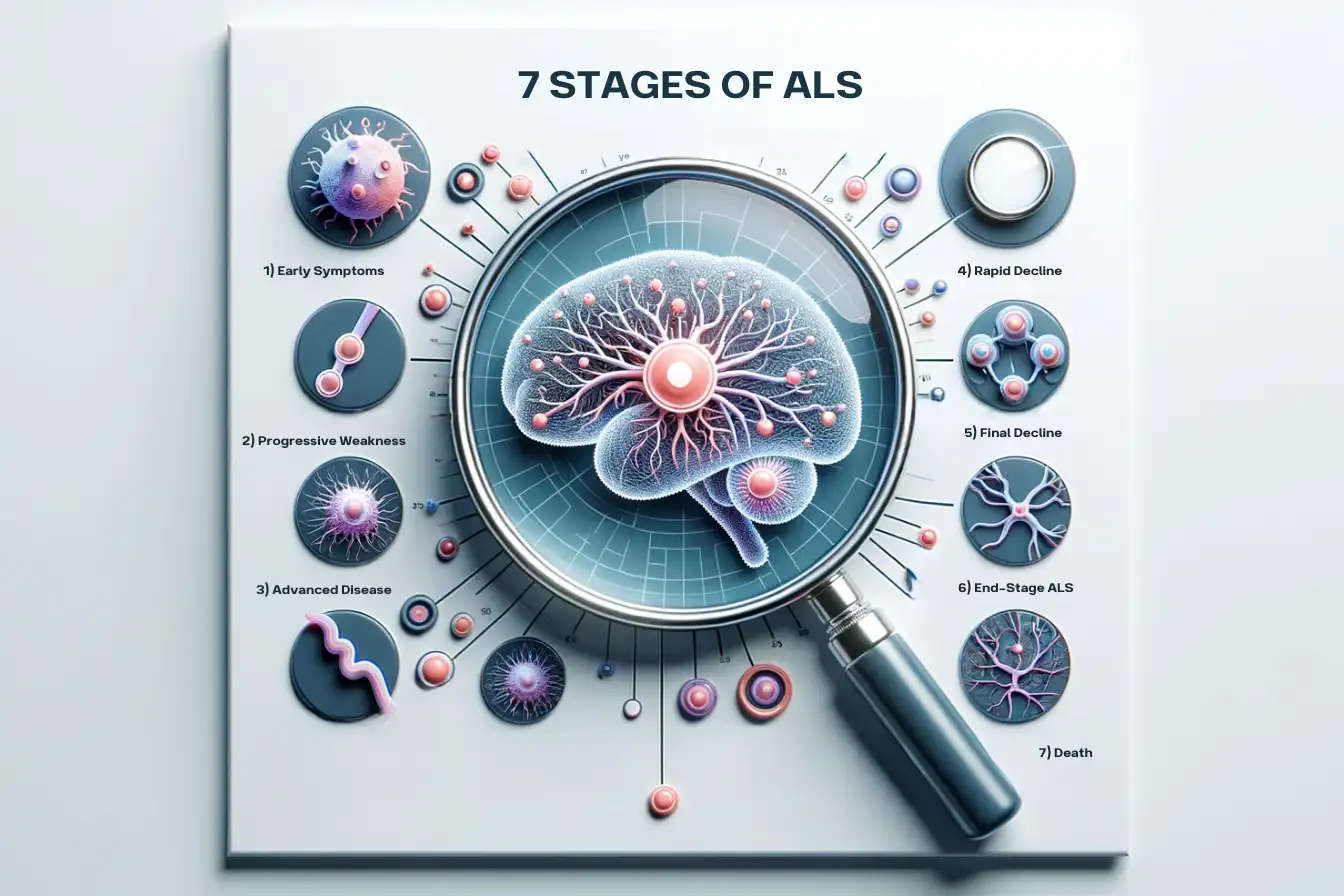
The 7 Stages of ALS
The journey through Amyotrophic Lateral Sclerosis (ALS) unfolds through distinct stages, each representing a different phase of the disease and its impact on the individual. Understanding these stages provides insight into the progression of ALS and helps in anticipating the care and support required at each phase. Below are the 7 stages of ALS detailed:
Early Symptoms Stage: The initial encounter with ALS often comes with subtle symptoms, which may include muscle weakness, twitching, cramping, or stiffness manifesting in areas such as the arms, legs, shoulders, or tongue. These symptoms are usually mild at the outset and may go unnoticed.
Progressive Weakness Stage: As the disease advances, the demise of motor neurons exacerbates muscle weakness and atrophy, which gradually spreads to other regions of the body. Everyday activities like walking, speaking, swallowing, and breathing become increasingly challenging.
Advanced Disease Stage: At this juncture, most muscles are severely weakened, leading to significant disability. Patients require assistance with self-care routines and often lose the ability to walk or stand independently. Challenges with breathing and swallowing become common occurrences.
Rapid Decline Stage The trajectory of decline varies among individuals, but eventually, most people with ALS necessitate constant care. Paralysis may extend to the limbs and trunk, and difficulties in breathing and swallowing intensify.
Final Decline Stage This stage witnesses widespread paralysis. The functional use of arms and legs diminishes, alongside the ability to speak and swallow. Breathing becomes severely impaired, necessitating the use of ventilators and feeding tubes.
End-Stage ALS: Patients transition into a state of complete paralysis, with the exception of eye movements. Permanent ventilator support becomes indispensable for breathing, and total loss of communication ensues.
Death: The final stage culminates in death, predominantly due to respiratory failure. On average, the survival span from onset to death ranges between 3-5 years, although about 10% of individuals endure for 10 years or more.
ALS Early Symptoms Stage
Very early ALS symptoms are often elusive and can vary significantly among individuals. It is crucial for patients and healthcare providers to recognize the early symptoms to ensure timely intervention and better management of the stages of lou gehrig's disease.
ALS Symptoms and Onset Variability
Muscle Weakness: One of the initial signs of ALS is muscle weakness, which may manifest in various ways such as difficulty in lifting objects, climbing stairs, or even walking.
Muscle Spasticity: Patients may experience stiffness in the muscles which can be discomforting.
Cramping and Fasciculations: Muscle cramping and twitching are common and can be quite painful.
Limb Onset vs. Bulbar Onset: ALS can have different onset types. Limb onset starts in the arms or legs, while bulbar onset begins in the bulbar muscles affecting speech and swallowing.
Symptoms | Description | Limb Onset Prevalence | Bulbar Onset Prevalence |
|---|---|---|---|
Muscle Weakness | Difficulty in performing routine tasks | 60-70% | 20-30% |
Muscle Spasticity | Stiffness in muscles | Common | Less Common |
Cramping | Painful muscle cramps | Often | Occasionally |
Fasciculations | Muscle twitching | Often | Occasionally |
Impact on Daily Tasks and Lifestyle
Early-stage ALS can subtly impact a person's ability to perform routine tasks. Over time, the muscle weakness can lead to:
Mobility Issues: Difficulty in walking or moving around.
Speech Challenges: Speaking may become slurred or nasal sounding, particularly in bulbar onset ALS.
Difficulty in Chewing and Swallowing: Bulbar onset ALS affects the muscles used in chewing and swallowing earlier than limb onset.
Diagnosing ALS
Diagnosing ALS is a meticulous process as there is no definitive test for it. An accurate diagnosis is crucial for devising an appropriate management plan.
Every ALS patient's journey is unique, which necessitates a tailored therapeutic approach for each individual. By leveraging advanced diagnostic tools and personalized medicine techniques, it's possible to design a more patient-centered treatment plan. This can include a combination of physical therapy, occupational therapy, speech therapy, nutritional guidance, and personalized medication management, all aimed at enhancing the quality of life and slowing down the disease progression as much as possible.
Challenges in Diagnosis
Lack of Clear Markers: ALS diagnosis is often challenging due to the lack of clear biological markers.
Symptom Overlap: Symptoms of ALS may overlap with other neurodegenerative disorders, making it difficult to diagnose.
Diagnostic Tests and Procedures
Several tests and procedures are involved in diagnosing ALS. These may include:
Blood and Urine Tests: To rule out other conditions.
Electromyography (EMG): Evaluates the electrical activity of muscles.
Nerve Conduction Study (NCS): Measures the speed of electrical signals through the nerves.
Magnetic Resonance Imaging (MRI): While it cannot diagnose ALS, it can rule out other conditions that may mimic ALS symptoms.
Test | Purpose | Description |
|---|---|---|
Blood and Urine Tests | Rule out other conditions | Checks for specific markers to eliminate other diseases |
EMG | Evaluate muscle electrical activity | Measures the electrical activity produced by muscles |
NCS | Assess nerve functionality | Evaluates the speed and strength of signals traveling between nerves |
MRI | Rule out other conditions | Uses magnetic fields to create detailed images of the brain and spinal cord |
Importance of Early Diagnosis
Early diagnosis of ALS allows for:
Better Management: Early intervention can help manage symptoms better and improve the quality of life.
Informed Decision Making: Patients and families can make informed decisions regarding treatment options and planning for the future.
Middle-Stage ALS
As ALS progresses into its middle stages, the symptoms experienced during the early stages tend to worsen. The loss of muscle control and strength becomes more widespread, impacting not only the individual’s mobility but also their ability to perform daily tasks independently.
The advent of technology has brought about a plethora of assistive devices and communication aids that can significantly improve the quality of life for individuals in the middle stages of ALS. Innovations like eye-tracking software for communication, smart home adaptations, and wearable devices to monitor physiological parameters can empower individuals with ALS to maintain a level of independence and engage with their surroundings and loved ones.
Worsening Symptoms
Muscle Atrophy: The muscles continue to weaken and shrink in size.
Increased Weakness: The weakness spreads to other parts of the body, making daily activities more challenging.
Paralysis: Some muscles may become paralyzed, restricting movement further.
Impact on Mobility and Daily Living
The debilitating symptoms of middle-stage ALS significantly affect a person’s life:
Mobility: Walking becomes more difficult, and individuals may require aids like walkers or wheelchairs.
Daily Activities: Tasks like dressing, eating, and personal hygiene become challenging and may require assistance.
Communication: Speech may become more slurred, and alternative communication methods might be necessary.
Emotional and Psychological Adjustments
The progressive loss of independence and the increasing need for assistance often lead to emotional and psychological challenges. It's crucial for individuals and their families to have access to supportive resources to cope with these changes.
Mandatory Gastrostomy Stage ALS
As swallowing becomes increasingly difficult, maintaining proper nutrition becomes a challenge. The mandatory gastrostomy stage addresses this issue by the introduction of a feeding tube to ensure adequate nutrition and hydration.
Feeding Tube Placement
Procedure: A gastrostomy tube (G-tube) is placed directly into the stomach to deliver nourishment and fluids.
Benefits: Ensures adequate nutrition, hydration, and medication administration.
AspectDescriptionProcedurePlacement of a gastrostomy tube (G-tube)BenefitsAdequate nutrition, hydration, and medication administration
Nutritional Management
Proper nutritional management is crucial in maintaining the health and well-being of individuals with ALS.
Dietary Planning: Tailoring diets to meet individual needs and preferences.
Monitoring: Regular monitoring by healthcare providers to ensure nutritional needs are met.
Late Stage ALS
In the late stages of ALS, most voluntary muscles become paralyzed. The muscles involved in breathing, speaking, and swallowing are severely affected, leading to significant challenges.
Respiratory Challenges
Breathing Difficulties: Breathing becomes laborious as the respiratory muscles weaken.
Ventilator Support: Individuals may require ventilator support to aid in breathing.
Mobility and Communication Challenges
Severe Mobility Limitations: Movement becomes extremely limited, and individuals become bedridden.
Communication Barriers: Speaking becomes almost impossible, necessitating alternative communication methods.
Final Stages of ALS
The final stages of ALS are characterized by complete paralysis, and most individuals will require full-time care and support. The focus during this stage shifts to providing comfort and ensuring a dignified quality of life as much as possible.
End-of-Life Care
Hospice Care: Provides comfort, pain management, and support to individuals and their families.
Respiratory Failure: The most common cause of death in ALS patients, requiring careful management and support.
The global ALS community, comprising of medical professionals, researchers, patients, and caregivers, has been instrumental in driving awareness, fundraising for research, and providing support for those affected by ALS. Various initiatives like the Ice Bucket Challenge have not only raised substantial funds for ALS research but also fostered a sense of global solidarity towards finding a cure. In light of this, it's crucial for individuals and families affected by ALS to connect with local and global ALS communities for support, advocacy, and staying updated on the latest research and treatment advancements.
Medication and Support
Medication: Medications to relieve discomfort, anxiety, and other symptoms.
Supportive Care: Ensuring a comfortable and dignified experience through supportive care and symptom management.
Conclusion
The trajectory of Amyotrophic Lateral Sclerosis (ALS) through its seven stages portrays a profound impact on the affected individuals and their loved ones. From the onset of mild symptoms to a state of complete paralysis, the progressive nature of ALS necessitates a comprehensive understanding, early diagnosis, and a multidisciplinary approach to care and management. By delving into each stage, we can better fathom the evolving needs of individuals with ALS, thus paving the way for enhanced patient-centric care, support, and eventually, more refined therapeutic interventions.
Stem Cells and ALS
In the realm of ALS research, stem cell therapy emerges as a beacon of hope. Stem cells, with their ability to differentiate into various cell types, offer a potential pathway to treat or even reverse the damage caused by ALS. Scientists are exploring the transplantation of stem cells to replace the damaged motor neurons or to provide a supportive environment for the remaining neurons to function optimally.
Numerous studies and clinical trials are underway to ascertain the safety and efficacy of stem cell therapies in ALS. The promise of stem cells lies not only in halting the progression of ALS but also in unveiling the mysteries of this devastating disease, thereby fostering the development of more effective treatments in the near future.
Frequently Asked Questions
How do you know the end is near with ALS?
ALS is a progressive neurodegenerative disease, so there is no single point that definitively indicates the end of life. However, there are several signs that suggest the later stages of ALS:
Severe muscle weakness and paralysis, affecting ability to move, speak, swallow, and breathe.
Weight loss and muscle wasting from dysphagia and poor nutrition.
Recurrent pneumonia or respiratory infections.
Marked decline in respiratory function tests and forced vital capacity.
Increasing difficulty clearing secretions and shortness of breath.
Cognitive and behavioral changes like frontotemporal dementia.
Failure to thrive and fatigue despite maximal therapy.
As ALS progresses, patients eventually require mechanical ventilation to breathe. At this point, along with significant weight loss, recurrent infections, and paralysis, hospice enrollment is typically recommended.
What is the death stage of ALS?
The terminal or end stage of ALS is characterized by:
Near complete paralysis and inability to move or speak.
Severe dysphagia requiring feeding tube for nutrition.
Respiratory failure requiring continuous ventilator support.
Recurrent pneumonia and respiratory infections.
Marked weight loss and muscle wasting.
Minimal communication through eye movements.
In the final weeks, patients are bedbound, unable to eat or breathe independently, and require around-the-clock care. Death usually occurs due to respiratory failure.
How fast do you deteriorate with ALS?
The rate of progression and deterioration varies significantly in ALS. On average:
Life expectancy after diagnosis is 2-5 years.
25% of patients survive 5 years after diagnosis.
10% of patients have a more slowly progressing form and survive 10 years or more.
Factors influencing disease progression include:
Age at onset - faster progression with older age of onset.
Site of onset - bulbar onset progresses more rapidly.
Genetics - some gene mutations have faster progression.
Though variable, progression is generally relentless with gradual worsening over time. Patients lose about 5% of remaining motor function per month.
What are the 7 stages of ALS?
There is no standard staging system for ALS, but one proposed system identifies 6 stages:
Symptom onset
Diagnosis
Independence
Assisted independence
Moderate loss of function
Advanced loss of function
Death
The last 2 stages show severe impairment and align with end stages described above.
When should an ALS patient go on hospice?
Guidelines recommend hospice referral when:
Patient exhibits critically impaired breathing capacity (forced vital capacity < 30%)
Patient has significantly impaired swallowing and poor nutrition
Patient shows rapidly progressing muscle weakness and functional decline
Additional considerations include:
Patient desires comfort-focused care over life prolonging interventions
Patient develops recurrent pneumonia or respiratory infections
Patient exhibits marked weight loss and is bedbound
Early hospice enrollment allows patients to prepare emotionally and practically for end of life.
When should I start hospice for ALS?
The optimal time to enroll in hospice care is when the patient:
Has severe breathing or swallowing impairment impacting quality of life
No longer responds meaningfully to ALS-slowing medication
Needs assistance with most activities of daily living
Wishes to focus on comfort rather than prolonging life
Guidelines recommend starting discussions about hospice 6-12 months after diagnosis to allow time to prepare. Hospice can be started earlier for patients with aggressive ALS.
What are the two most common causes of death in ALS patients?
The two leading causes of death for ALS patients are:
Respiratory failure - inability to breathe due to muscle weakness. This accounts for about 80% of ALS deaths.
Pneumonia - lung infection often due to aspiration or impaired cough. Pneumonia accounts for 10-15% of ALS deaths.
Other less common causes include pulmonary embolism, malnutrition, and arrhythmias. Ultimately most patients die from respiratory-related complications.
Is ALS painful at the end?
ALS itself does not cause pain, as it damages motor neurons rather than sensory nerves. However, several factors can lead to pain at end stages:
Muscle cramps from fasciculations
Joint stiffness or skin sores from immobility
Muscle spasms or contractions
Complications like pneumonia, blood clots, or bed sores
Good palliative care focusing on comfort can help manage pain at the end stages.
Why is oxygen bad for ALS patients?
Oxygen supplementation provides no benefit and may actually be harmful in ALS patients with respiratory insufficiency. Reasons include:
It removes the drive to breathe, leading to carbon dioxide retention
It can suppress respiratory function and hasten the need for mechanical ventilation
It may increase oxidative damage and worsen ALS progression
Oxygen should be avoided unless the patient has hypoxemia or a low oxygen saturation.
What are 3 things that can lead to ALS?
The causes of ALS are not fully understood, but three major factors believed to contribute to ALS risk include:
Genetics - About 10% of cases are inherited. Mutations in genes like SOD1, C9ORF72, TARDBP, and FUS can cause familial ALS.
Environment - Smoking, heavy metals, pesticides, trauma, and viruses may increase ALS risk.
Age - Most cases occur in people 40-70 years old. Age-related changes like oxidative damage may promote ALS.
Other proposed factors include glutamate excitotoxicity, protein misfolding, and mitochondrial dysfunction. The cause likely involves a complex interaction between genetic and environmental triggers.
Can ALS go into remission?
True remission, where the disease completely stops progressing, has not been observed in ALS. However, some patients experience temporary plateaus where progression stalls for a period before worsening again.
In rare cases, patients may exhibit neuromuscular recovery and regain certain functions after an initial diagnosis of ALS. But ALS is considered a progressive disease that continues advancing without long-term remission.





Post comments