
by Kimm Fesenmaier, California Institute of Technology
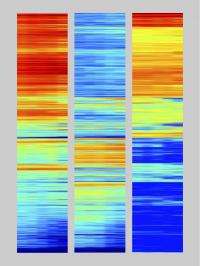
These bars summarize the epigenetic markers correlated with RNA-expression levels for each of about 20,800 known genes in the mouse genome. The left column shows epigenetic markers correlated with activation, the middle shows repression markers, and the right shows the RNA levels expressed by the corresponding genes. Reading across each row, genes that change expression or epigenetic markers during T-cell development change color. Red denotes the highest level of epigenetic marking or RNA expression, while blue denotes the lowest. Credit: Zhang/Rothenberg/Caltech
What happens to a stem cell at the molecular level that causes it to become one type of cell rather than another? At what point is it committed to that cell fate, and how does it become committed? The answers to these questions have been largely unknown. But now, in studies that mark a major step forward in our understanding of stem cells' fates, a team of researchers from the California Institute of Technology (Caltech) has traced the stepwise developmental process that ensures certain stem cells will become T cells—cells of the immune system that help destroy invading pathogens.
"This is the first time that a natural developmental process has been dissected in such detail, going from step to step to step, looking at activities of all the genes in the genome," says Ellen Rothenberg, the principal investigator on the study and the Albert Billings Ruddock Professor of Biology at Caltech. "It means that in genetic terms, there is virtually nothing left hidden in this system."
The study was led by Jingli A. Zhang, a graduate student in Rothenberg's lab, who is now a postdoctoral scholar at Caltech. The group's findings appear in the April 13 issue of the journal Cell.
The researchers studied multipotent hematopoietic precursor cells—stem-cell-like cells that express a wide variety of genes and have the capability to differentiate into a number of different blood-cell types, including those of the immune system. Taking into consideration the entire mouse genome, the researchers pinpointed all the genes that play a role in transforming such precursor cells into committed T cells and identified when in the developmental process they each turn on. At the same time, the researchers tracked genes that could guide the precursor cells to various alternative pathways. The results showed not only when but also how the T-cell-development process turned off the genes promoting alternative fates.
"We were able to ask, 'Do T-cell genes turn on before the genes that promote some specific alternative to T cells turn off, or does it go in the other order? Which genes turn on first? Which genes turn off first?'" Rothenberg explains. "In most genome-wide studies, you rarely have the ability to see what comes first, second, third, and so on, in a developmental progression. And establishing those before-after relationships is absolutely critical if you want to understand such a complicated process."
The researchers studied five stages in the cascade of molecular events that yields a T cell—two before commitment, a commitment stage, and then two following commitment. They identified the genes that are expressed throughout those stages, including many that code for regulatory proteins, called transcription factors, which turn particular genes on or off. They found that a major regulatory shift occurs between the second and third stages, when T-cell commitment sets in. At that point, a large number of the transcription factors that activate genes associated with uncommitted stem cells turn off, while others that activate genes needed for future steps in T-cell development turn on.
The researchers looked not only at which genes are expressed during the various stages but also at what makes it possible for those genes to be expressed at that particular time. One critical component of regulation is the expression of transcription-factor genes themselves. Beyond that, the researchers were interested in identifying control sequences—the parts of genes that serve as docking sites for transcription factors. These sequences are often very difficult to identify in mice and humans using classical molecular-biology techniques; scientists have spent as many as 10 years trying to create a comprehensive map of the control sequences for a single gene.
To create a map of likely control sequences, Zhang studied epigenetic markers. These are chemical modifications, such as those that change the way the DNA is bundled. They become associated with particular regions of DNA as a result of the action of transcription factors and can thereby affect how easy or hard it is for a neighboring gene to be turned on or off. By identifying DNA regions where epigenetic markers are added or removed, Rothenberg's group has paved the way for researchers to identify control sequences for many of the genes that turn on or off during T-cell development.
In some ways, Rothenberg says, her team is taking a backward approach to the problem of locating these control sequences. "What we're saying is, if we can tell that a gene is turned on at a certain point in terms of producing RNA, then we should also be able to look at the DNA sequences right around it and ask, 'Is there any stretch of DNA sequence that adds or loses epigenetic markers at the same time?'" Rothenberg says. "If we find it, that can be a really hot candidate for the control sequences that were used to turn that gene on."
Two methodologies have made it possible to complete this work. First, ultra-high-throughput DNA sequencing was used to identify when major changes in gene expression occur along the developmental pathway. This technique amplifies DNA sequences taken throughout millions of cell samples, puts all of the bits in order, compares them to the known genome sequence (for mice, in this case), and identifies which of the various genes are enriched, or found in greater numbers. Those that are enriched are the ones most likely to be expressed. The team also used a modified version of this sequencing technique to identify the parts of the genome that are associated with particular epigenetic markers. Coauthor Barbara Wold, Caltech's Bren Professor of Molecular Biology, is an expert in these so-called "next-generation" deep-sequencing technologies and provided critical inspiration for the study.
A second important methodology involved an in vitro tissue-culture system developed in the lab of Juan Carlos Zúñiga-Pflücker of the University of Toronto, which enabled the Caltech researchers to mass-produce synchronized early T-cell precursors and to see the effect of altered conditions on individual cells in terms of producing T cells or other cells.
More information: "Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity," Cell (2012).
Journal information: Cell






Post comments