
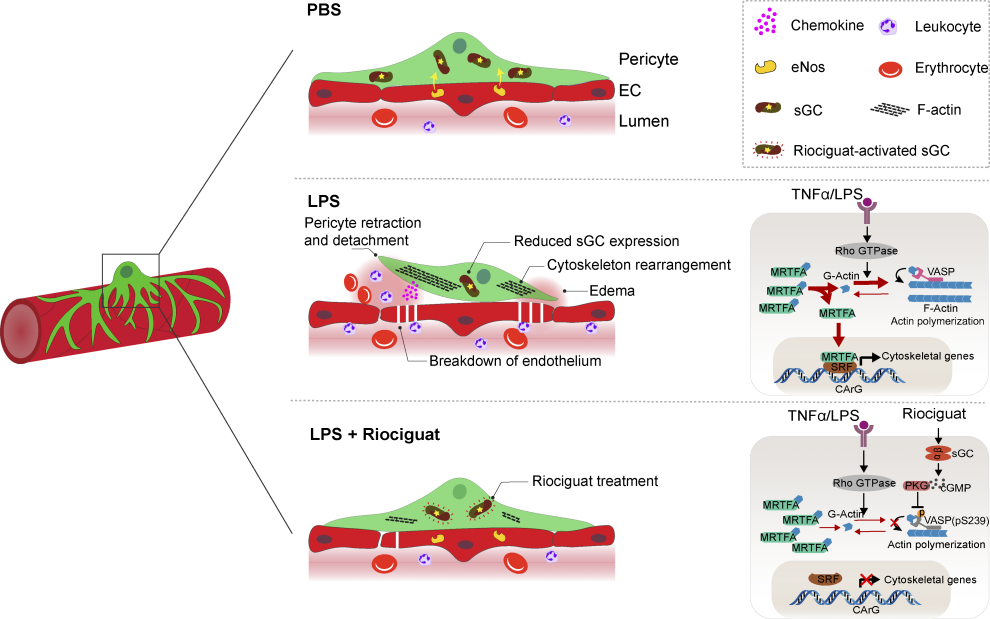
Working model. eNOS–sGC signaling, as a key mediator of EC–pericyte crosstalk, controls vascular integrity. In LPS-induced ALI, the eNOS–sGC crosstalk is significantly reduced, resulting in cytoskeleton rearrangement, pericyte detachment, and elevated chemokine expression. This leads to immune cell infiltration, breakdown of the endothelium, and lung edema. Administration of the sGC stimulator Riociguat is sufficient to stabilize EC–pericyte interaction, promoting vascular integrity and reducing inflammation-induced lung injury.
Credit:DOI:10.1084/jem.20211422
Cellular crosstalk is a fundamental mechanism that underlies the complex interactions between different cell types within the cardiovascular system. This communication is crucial for maintaining homeostasis, orchestrating developmental processes, and responding to pathological stimuli. For instance, in the pulmonary vasculature, endothelial cells and pericytes were found to coordinate their activities to maintain vascular integrity and regulate inflammation. Disruption of these interactions, as observed in acute lung injury (ALI), resulted in increased vascular permeability and lung injury.
ALI and its more severe form, acute respiratory distress syndrome (ARDS), are life-threatening conditions characterized by increased vascular permeability and subsequent lung edema and inflammation. While numerous therapeutic strategies have been explored, their efficacy remains limited. The study conducted by He et al. sheds light on a novel therapeutic target within the vascular niche that could potentially improve outcomes in ALI patients.
The research team employed a comparative genome-wide analysis of endothelial cell (EC) and pericyte interactions in the lungs of healthy mice and mice with LPS-induced ALI. They identified a significant impairment in the endothelial nitric oxide (NO) and soluble guanylate cyclase (sGC) signaling pathway in the lungs of ALI mice. This impairment was associated with pericyte detachment, loss of endothelial integrity, and increased infiltration of inflammatory cells.
To investigate the therapeutic potential of restoring NO-sGC signaling, the researchers administered the sGC stimulator Riociguat to LPS-treated mice. Surprisingly, Riociguat alone did not significantly alter vascular integrity. However, in combination with LPS, Riociguat effectively reduced Evans blue leakage, lung edema, and the infiltration of inflammatory cells, demonstrating its protective effects against ALI-induced lung injury.
Further investigation revealed that the beneficial effects of Riociguat were primarily attributed to its activation of pericyte sGC. In contrast, inactivating sGC in vascular smooth muscle cells (vSMCs) or platelets did not compromise Riociguat's protective effects. This suggested that pericytes play a pivotal role in maintaining vascular integrity and that activating sGC in pericytes is a promising therapeutic strategy for ALI.
The researchers conducted in vitro experiments using microfluidic chips to further elucidate the mechanisms by which NO-sGC signaling protects against ALI. They found that TNFα stimulation induced pericyte retraction and detachment, leading to increased vascular leakage. However, activating sGC signaling with cGMP reversed these morphological changes and preserved pericyte coverage, demonstrating the critical role of sGC in maintaining pericyte stability and vascular integrity.
The study also explored the molecular mechanisms underlying the protective effects of NO-sGC signaling. They found that Riociguat treatment suppressed the expression of cytoskeletal genes in pericytes, leading to reduced F-actin formation and MRTFA/SRF-dependent de novo synthesis of genes associated with cytoskeleton rearrangement. This suppression ultimately resulted in the stabilization of EC-pericyte interactions and preserved the integrity of the alveolar vasculature.
Additionally, the researchers observed that Riociguat treatment reduced LPS-induced inflammatory cytokines and chemokines expression in lung pericytes. This suggests that NO-sGC signaling may exert anti-inflammatory effects by regulating the expression of inflammatory mediators.
Overall, this study provides compelling evidence that impaired NO-sGC crosstalk in the vascular niche contributes to increased vascular permeability and lung injury in ALI. The successful demonstration of Riociguat's protective effects against ALI, primarily through its activation of pericyte sGC, opens up a promising therapeutic avenue for the treatment of this devastating condition.
Similarly, in the context of cardiac remodeling, Chen et al. revealed that fibroblasts and cardiomyocytes engaged in bidirectional signaling to regulate extracellular matrix (ECM) deposition, myocyte growth, and contractility. This interplay was tightly regulated and disrupted in diseases like heart failure (HF), leading to excessive fibrosis and hypertrophy.
This study investigated the role of Integrin beta-like 1 (ITGBL1) in cardiac remodeling and HF by examining its expression and function in both in vitro cell experiments and in vivo mouse models of pressure overload-induced hypertrophy.
The researchers found that ITGBL1 was highly expressed in cardiac fibroblasts (CFs) compared to cardiomyocytes (CMs), and its expression was further upregulated in CFs under pro-fibrotic stimuli like angiotensin-II (AngII) or phenylephrine.
Overexpression of ITGBL1 in CFs promoted their activation, as evidenced by increased expression of fibrotic markers like α-SMA and collagen, enhanced migration, contraction, and proliferation. Conversely, knockdown of ITGBL1 inhibited AngII-induced CF activation.
ITGBL1 competed with the metastasis suppressor NME1 for binding to the TGF-β receptor-associated protein STRAP, thereby impairing the suppressive effect of the STRAP-NME1 complex on TGF-β signaling. This resulted in increased phosphorylation of Smad2/3 and further activation of CFs.
Additionally, Conditioned medium from CFs overexpressing ITGBL1 induced hypertrophy in CMs, characterized by increased expression of hypertrophic markers like ANP and BNP, and enlarged cell size with more organized sarcomere assembly. Knockdown of ITGBL1 in CFs inhibited this pro-hypertrophic effect.
Furthermore, ITGBL1 activated the Wnt/β-catenin signaling pathway in CMs, leading to increased expression of Wnt-related genes and downstream targets involved in cell proliferation and hypertrophy. Additionally, ITGBL1 also affected the phosphorylation of ERK1, suggesting involvement of the MAPK/ERK pathway in CM hypertrophy.
Using adeno-associated virus (AAV) to specifically knock down ITGBL1 in CFs in vivo, the researchers found that ITGBL1 knockdown significantly improved cardiac function and reduced cardiac hypertrophy and fibrosis in a mouse model of transverse aortic constriction (TAC).
In summary, this study provided compelling evidence that ITGBL1 was a novel and important mediator of cardiac remodeling and HF. ITGBL1 promoted CF activation through the ITGBL1-NME1-TGF-β-Smad2/3 pathway and induced CM hypertrophy through the Wnt/β-catenin pathway. These findings suggested that ITGBL1 could be a potential therapeutic target for preventing and treating cardiac remodeling and HF. Understanding the specific signaling pathways and molecular mechanisms involved in cellular crosstalk is essential for developing targeted therapies that restore normal function and prevent the progression of cardiovascular diseases.
Reference:
He H, Yang W, Su N, Zhang C, Dai J, Han F, Singhal M, Bai W, Zhu X, Zhu J, Liu Z, Xia W, Liu X, Zhang C, Jiang K, Huang W, Chen D, Wang Z, He X, Kirchhoff F, Li Z, Liu C, Huan J, Wang X, Wei W, Wang J, Augustin HG, Hu J. Activating NO-sGC crosstalk in the mouse vascular niche promotes vascular integrity and mitigates acute lung injury. J Exp Med. 2023 Feb 6;220(2):e20211422.
Chen X, Li X, Wu X, Ding Y, Li Y, Zhou G, Wei Y, Chen S, Lu X, Xu J, Liu S, Li J, Cai L. Integrin beta-like 1 mediates fibroblast-cardiomyocyte crosstalk to promote cardiac fibrosis and hypertrophy. Cardiovasc Res. 2023 Aug 19;119(10):1928-1941.




Post comments