
Credit:Raising NAD Level Stimulates Short-Chain Dehydrogenase/Reductase Proteins to Alleviate Heart Failure Independent of Mitochondrial Protein Deacetylation+.
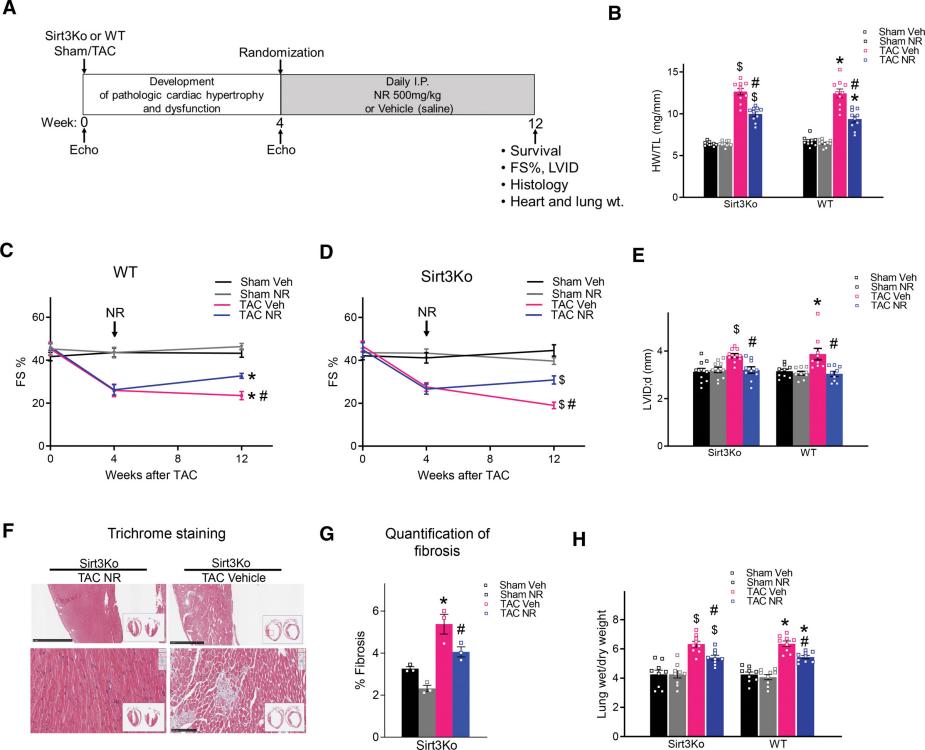
Increasing NAD level blunted the development of heart failure in Sirt3-deficient mice. A+, Experimental protocol. At 4 weeks after transverse aortic constriction (TAC), mice were treated with nicotinamide riboside chloride (NR) or vehicle (saline) for 8 weeks. B, Ratio of heart weight/tibia length (HW/TL) was measured after 8 weeks of treatment. C and D, Fractional shortening (FS) for WT and Sirt3Ko mice was quantified by echocardiography. E, Left ventricular (LV) dilation (left ventricular internal diameter at end diastole [LVID;d]) quantified by echocardiography for WT and Sirt3Ko mice. F, Representative LV cross-sections of Sirt3Ko TAC with or without NR treatment stained with Masson’s trichrome. Scale bars, top, 1 mm; and bottom, 100 μM. G, Quantification of fibrotic area. H, Lung edema (wet/dry lung weight) was measured after 8 weeks of treatment. All data are presented as mean±SEM. P value was calculated by mixed-effects analysis followed by Tukey post hoc analysis (C and D) or 2-way ANOVA followed by Tukey post hoc analysis for B, E, G, and H. n≥5 per group except G (n=3). *P<0.05 vs WT sham veh, $P<0.05 vs Sirt3Ko sham veh, and #P<0.05 vs TAC veh. NAD indicates oxidized nicotinamide adenine dinucleotide; Sirt3, sirtuin 3; Sirt3Ko, sirtuin 3 knockout; veh, vehicle; and WT, wild-type.+
In heart failure, mitochondrial dysfunction in cardiomyocytes is a key pathological mechanism. This dysfunction not only results in insufficient energy production, which prevents cardiomyocytes from maintaining normal function, but also triggers oxidative stress, inflammatory responses, and apoptosis, further exacerbating the progression of heart failure. NAD+(Nicotinamide adenine dinucleotide, NAD) is a crucial regulator of mitochondrial function. While previous studies have demonstrated that increasing NAD+ levels can improve heart failure, the specific molecular mechanisms remain unclear. Particularly, the pathways and proteins through which NAD+ exerts its effects require further investigation. Matthew et al. utilized Sirt3-deficient and wild-type mouse models, induced cardiac dysfunction through pressure overload, and then compared the effects of NR (nicotinamide riboside, NR) treatment. This study aimed to explore whether increasing NAD+ levels can ameliorate heart failure by activating the SDR (short-chain dehydrogenase/reductase, SDR) family proteins and whether this process is independent of mitochondrial protein deacetylation, primarily mediated by Sirt3.
Researchers employed TAC (transverse aortic constriction, TAC) surgery to increase cardiac pressure by constricting the aorta in mouse models, thereby inducing cardiac hypertrophy and heart failure. Mice were divided into three groups: WT (wild-type, WT), Sirt3-deficient , and control. NR treatment was initiated four weeks post-surgery and continued for eight weeks. The experimental results showed that NR treatment significantly improved FS (fractional shortening, FS) in both WT and Sirt3 mice, indicating that NR treatment can attenuate the progression of heart failure. Additionally, NR treatment significantly reduced cardiac hypertrophy and fibrosis in both WT and Sirt3 mice and markedly increased the NAD+/NADH ratio in the hearts of these mice. Although NR treatment decreased mitochondrial protein acetylation in WT mice, there was no significant change in Sirt3 mice. RNA sequencing analysis revealed that NR treatment upregulated genes related to mitochondrial metabolism, including those involved in the mitochondrial ETC (electron transport chain, ETC) and fatty acid β-oxidation. Finally, they measured the dehydrogenase activity of MRPP2 through enzymatic reactions, and the results showed that NR treatment restored MRPP2 dehydrogenase activity in both WT and Sirt3 mice. Through these detailed experimental designs and results, the study confirmed that increasing NAD+ levels can improve heart failure by activating SDR proteins, and this mechanism remains effective even in the absence of Sirt3.
Traditional NAD+ therapies primarily focus on activating Sirt3, which is believed to improve mitochondrial function by deacetylating mitochondrial proteins. However, the efficacy of Sirt3-mediated therapies has been inconsistent across different models, and its mechanisms are not entirely clear in some cases. This study, by increasing NAD+ levels, has revealed the importance of the SDR family proteins in regulating mitochondrial function, providing an alternative regulatory pathway apart from Sirt3. The research found that increasing NAD+ levels significantly improved heart failure regardless of the presence of Sirt3. This indicates that such therapy might be effective across different genetic backgrounds, offering broader applicability. Additionally, through detailed molecular biological analyses, the study elucidated the specific role of MRPP2 in NAD+ regulation and demonstrated the direct association between impaired MRPP2 function and heart failure. This provides a clear target for the development of future targeted therapies.
Although the paper has made significant progress in treating heart failure by increasing NAD+ levels, there are still some shortcomings and gaps from a critical perspective. For instance, the genetic background used in the study is relatively homogeneous. Future research should include mouse models with more diverse genetic backgrounds to comprehensively understand the effects of NAD+ enhancement. Additionally, the study used a fixed dose of NR and did not explore the impact of different doses, administration routes, and frequencies on the therapeutic effect. Furthermore, the experiments primarily focused on short-term effects, and the long-term effects of increasing NAD+ levels remain unclear. Future research should not only address these issues but also delve deeper into the mechanisms, such as the functions of SDR proteins in different cells and tissues, and other pathways through which NAD+ levels can be increased and their potential molecular mechanisms.
In summary, the study confirms that increasing NAD+ levels can improve heart failure by activating the SDR family proteins, particularly MRPP2. This discovery provides new insights and methods for treating heart failure and, compared to traditional Sirt3-mediated NAD+ therapies, offers broader applicability and potential clinical prospects. With further research, this finding could lead to the development of various new technologies, including novel NAD+ precursors, precise targeted drugs, personalized medical and diagnostic tools, and large-scale clinical trials. These advancements will not only help address the treatment challenges of heart failure but also promote the development of related industries, bringing significant social and economic benefits.
Walker, Matthew A., et al. "Raising NAD+ Level Stimulates Short-Chain Dehydrogenase/Reductase Proteins to Alleviate Heart Failure Independent of Mitochondrial Protein Deacetylation." Circulation 148.25 (2023): 2038-2057.




Post comments