

Credit:www.frepik.com
Heart failure is a leading cause of death worldwide, with approximately half of heart failure patients exhibiting preserved ejection fraction (HFpEF). The incidence of HFpEF is increasing due to rising rates of obesity, diabetes, and an aging population. However, effective treatment strategies for HFpEF are currently lacking, primarily because its pathophysiological mechanisms remain unclear. Research indicates that oxidative stress not only affects myocardial cell function but also leads to mitochondrial dysfunction and metabolic disturbances, which are closely related to HFpEF. The overexpression of inducible nitric oxide synthase (iNOS) produces excessive nitric oxide (NO), leading to oxidative stress and cellular damage. Studies have shown that iNOS inhibitors can significantly reduce oxidative stress and improve myocardial function in certain cardiovascular disease models. Additionally, previous research has found that iNOS can inhibit Akt activity through S-nitrosylation modification, leading to impaired insulin signaling and metabolic disturbances. Guo et al. evaluated the short-term and long-term effects of iNOS inhibition on cardiac and mitochondrial function, oxidative stress, and Akt S-nitrosylation by establishing an HFpEF mouse model and using pharmacological inhibition and gene knockout techniques.
Researchers first established an HFpEF mouse model using a high-fat diet (HFD) combined with Nx-nitro-L-arginine methyl ester (L-NAME). The model was then subjected to short-term intervention with the selective iNOS inhibitor L-NIL. Finally, iNOS knockout (iNOS-/-) mice were used to assess the long-term effects of the absence of iNOS. Results showed that in the HFpEF mouse model, HFD+L-NAME significantly increased iNOS expression levels. Short-term iNOS inhibition and iNOS gene knockout significantly improved diastolic function in HFpEF mice, as evidenced by the decreased E/A and E/E' ratios. This indicates that iNOS plays a crucial role in HFD+L-NAME-induced diastolic dysfunction and that inhibiting iNOS can improve the HFpEF phenotype. Both short-term iNOS inhibition and iNOS gene knockout reduced nitrate/nitrite levels in the urine of HFpEF mice and decreased Akt S-nitrosylation levels in myocardial tissue. This suggests that short-term iNOS inhibition and gene knockout are equally effective in reducing nitrosative stress and Akt S-nitrosylation. The phosphorylation of Akt at the Ser473 site was significantly reduced in myocardial tissue of HFpEF model mice, while iNOS inhibition restored Akt phosphorylation levels. Furthermore, in isolated cardiomyocytes, iNOS overexpression significantly inhibited insulin-mediated Akt phosphorylation and glucose uptake, whereas mutation of the Akt S-nitrosylation site significantly countered these adverse effects of iNOS. The expression of mitochondrial respiratory chain complexes (COX II, III, and IV) in myocardial tissue of HFpEF model mice was significantly reduced, along with significant decreases in citrate synthase activity and ATP levels. However, both iNOS inhibition and gene knockout improved mitochondrial function and ATP supply, indicating that iNOS inhibition can ameliorate the HFpEF phenotype by mitigating mitochondrial dysfunction. iNOS overexpression significantly increased NOX4 expression and superoxide levels in cardiomyocytes, reducing mitochondrial membrane potential. In contrast, L-NIL inhibition of iNOS alleviated these adverse effects, indicating that iNOS-induced oxidative stress and mitochondrial dysfunction play a significant role in HFpEF. Malondialdehyde (MDA) levels were significantly increased, and glutathione peroxidase (GSH-Px) activity was significantly reduced in myocardial tissue of HFpEF model mice. iNOS inhibition reduced excessive oxidative stress, increased Nrf2 phosphorylation, and upregulated HO-1 expression, suggesting that the Nrf2 pathway may mediate the protective effects of iNOS inhibition.
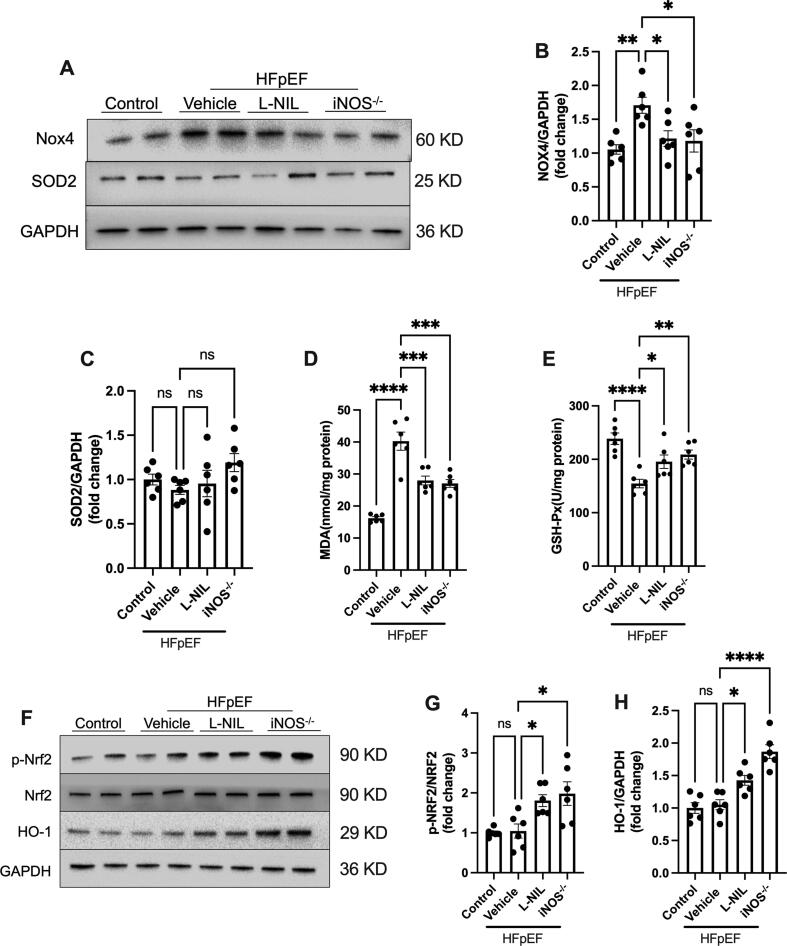
iNOS inhibition alleviated oxidative stress in the heart of HFpEF mice. (A-C) Representative immunostaining and semi-quantification of NOX4 and SOD2 in heart tissue samples. (D) Myocardial malondialdehyde levels. (E) GSH-Px activity. (F-H) Representative immunostaining and semi-quantification of p-Nrf2/Nrf2 as well as HO-1 in the heart tissue samples from mice in different experimental groups. n = 6. The data are shown as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s post hoc test. *, P < 0.05. **, P < 0.01. ***, P < 0.0005. ****, P < 0.0001. ns, no significant.
Credit:10.1016/j.jare.2022.03.003
In conclusion, this study confirms the critical role of iNOS as a pathophysiological driver in HFpEF. Short-term iNOS inhibition or gene knockout can improve mitochondrial function and reduce Akt S-nitrosylation, thereby alleviating the HFpEF phenotype. Given that selective iNOS inhibitors are safe and readily available, iNOS could be a potential therapeutic target for HFpEF.
Previous studies may have focused on a single mechanism or detection method, while this study increased its credibility by employing dual model validation. Through the comprehensive use of various methods, this research provides thorough and systematic evidence of the role of iNOS in HFpEF. Additionally, compared to observations at a single time point, this study offers a dynamic evaluation of iNOS's effects by employing both short-term and long-term iNOS inhibition.
Despite the significant contributions of this study to the research on iNOS and HFpEF, there are still some limitations. The study primarily relies on a mouse model, lacking validation across multiple animal models and human clinical data. Additionally, it does not thoroughly explore the specific roles of iNOS in different cardiac cell types or its interactions with other molecular pathways. Furthermore, the long-term safety and potential side effects of iNOS inhibition require further investigation.
Future directions could include the development of new drugs, optimization of treatment strategies, personalized medicine, improvement of drug delivery systems, and exploration of gene therapy. By advancing these areas in both research and clinical practice, we can potentially drive progress in the treatment of HFpEF and related diseases, providing more effective therapeutic options for patients.
Reference:
Guo Y, Wen J, He A, et al. iNOS contributes to heart failure with preserved ejection fraction through mitochondrial dysfunction and Akt S-nitrosylation[J]. Journal of advanced research, 2023, 43: 175-186.




Post comments