
In the intricate tapestry of biological systems, glucose metabolism, epigenetics, and disease development are inextricably linked, each influencing the other in a delicate dance that can lead to either health or pathology. Glucose, the primary fuel for cellular activities, is at the core of this interplay, with its metabolism shaping the epigenetic landscape and, in turn, the epigenetic modifications affecting glucose handling. Serio et al. employed a multifaceted approach, integrating chromatin immunoprecipitation sequencing (ChIP-seq), RNA sequencing (RNA-seq), and metabolomics to investigate the metabolic remodeling that occurs in the aging heart. Their findings revealed a critical role for enhancers, epigenetic regulators, and the transcriptional coactivator p300/CBP in promoting glycolysis and contributing to cardiac dysfunction.
The study began by examining the changes in histone modifications H3K27ac, H3K27me3, and H3K4me1 across the lifespan of mice. These modifications, which regulate the activity of promoters and enhancers, were found to dynamically reconfigure their genomic distribution as the heart ages. Specifically, H3K27ac levels increased, while H3K4me1 and H3K27me3 levels decreased, during the transition from adulthood to old age. This epigenetic shift was associated with distinct gene expression programs in aged hearts, characterized by the upregulation of genes involved in metabolism and cardiac hypertrophy.
To further understand the functional implications of these epigenetic changes, the authors identified a set of enhancers activated in aged cardiomyocytes. These enhancers were associated with genes involved in metabolism and heart disease, suggesting their role in driving the metabolic remodeling observed in aging hearts.
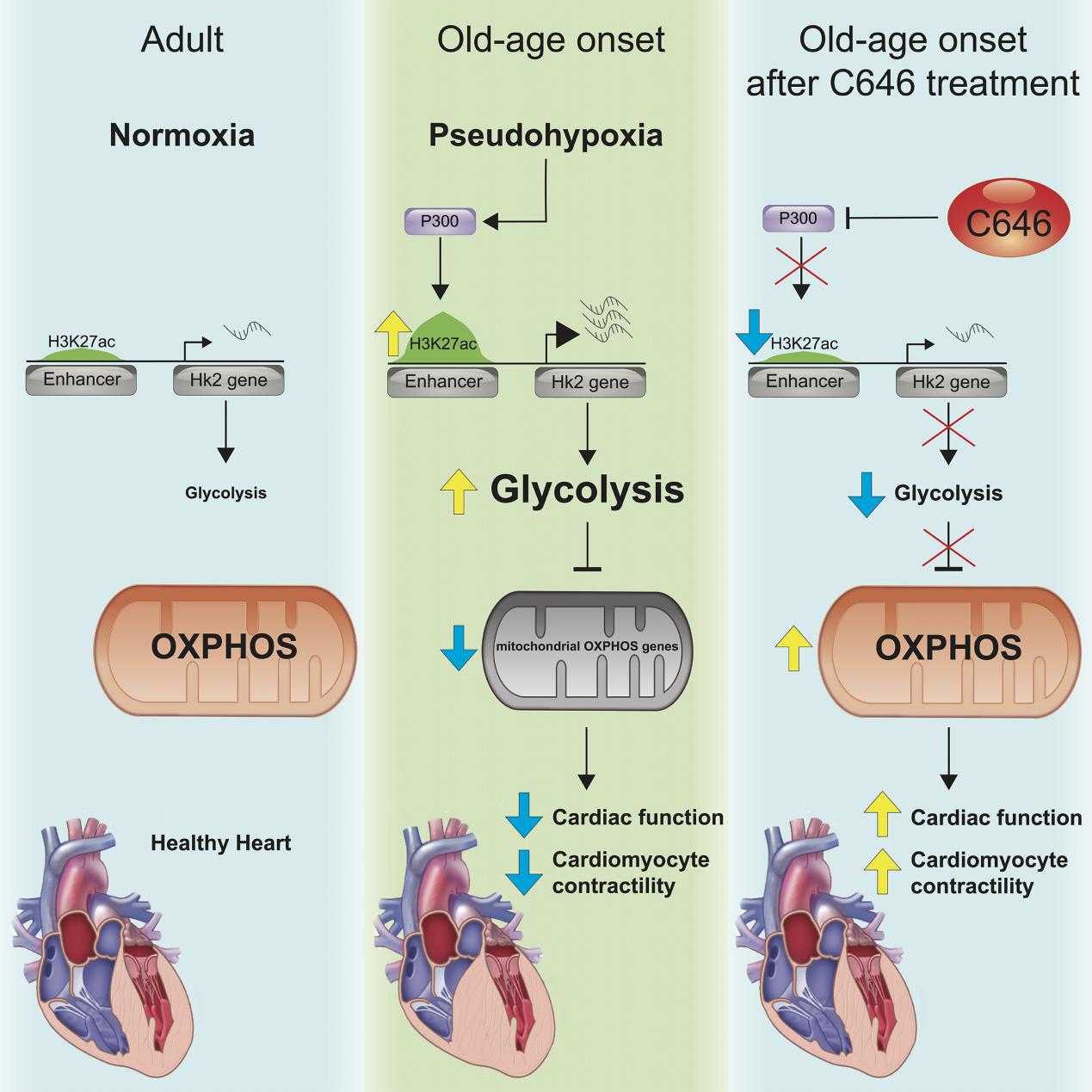
Further analysis revealed that the enhancers of key glycolysis genes, including Hk2, were activated during the onset of aging. This activation was linked to the metabolic remodeling characterized by increased glycolysis and decreased oxidative phosphorylation. The authors hypothesized that pseudohypoxia, a condition where hypoxia-related genes are expressed under normal oxygen levels, might be responsible for the activation of these enhancers and subsequent glycolytic switch.
The study focused on p300/CBP, a histone acetyltransferase known to activate enhancers and promote gene expression. p300/CBP was found to be crucial for the activation of the Hk2 enhancer during pseudohypoxia, suggesting its role in promoting glycolysis.
To test the functional significance of p300/CBP in cardiac aging, the authors pharmacologically inhibited this enzyme in aged mice. p300/CBP inhibition improved cardiac function, decreased Hk2 expression, and reversed the metabolic remodeling observed in aging hearts. These findings provided compelling evidence that p300/CBP-mediated enhancer activation was a key driver of age-related cardiac dysfunction.
This study provides valuable insights into the molecular mechanisms of cardiac aging and offers a potential therapeutic strategy for age-related HF. The authors propose that targeting enhancers and the epigenetic regulators that control their activity, such as p300/CBP, could be an effective approach to mitigate the decline in cardiac function associated with aging. Future studies are needed to validate the role of specific enhancers in cardiac aging and to investigate the pathways they regulate. Additionally, further research is required to explore the potential of p300/CBP inhibitors as a treatment for HF in humans.
Overall, this study represents a significant advance in our understanding of the molecular mechanisms of cardiac aging. The identification of p300/CBP as a key regulator of glycolysis and the potential therapeutic implications of its inhibition provide a promising avenue for the treatment of age-related HF.
While the first research has focused on the role of glucose metabolism in cardiac aging, the next study delved into the complex adaptations cancer cells undergo in response to glucose limitation. Saggese et al. investigated the impact of glucose restriction on lung adenocarcinoma (LUAD) cells and revealed a novel mechanism by which glucose deprivation induced dedifferentiation and enhances cell aggressiveness.
The authors employed a combination of in vitro and in vivo experiments, utilizing human LUAD cell lines, patient-derived organoids, and genetically engineered mouse models. They manipulated glucose concentrations in the culture media and employed various techniques such as Western blotting, RT-qPCR, RNA-seq, ChIP-seq, and metabolomics to analyze gene expression, protein levels, chromatin modifications, and metabolic changes.
It was observed that reduction of glucose levels in the culture media led to a downregulation of differentiation markers like FOXA2 and TTF1, while upregulating dedifferentiation markers like HMGA2 and GLUT1. This phenomenon was observed in both human and murine LUAD cells, suggesting a general mechanism of glucose-induced dedifferentiation in cancer cells.
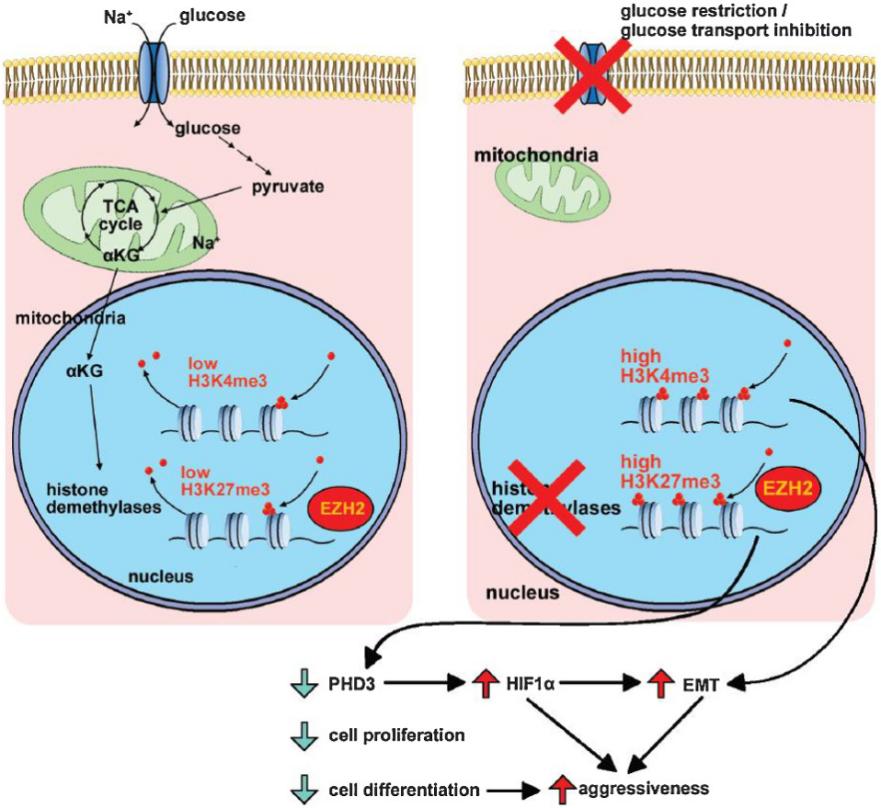
In addition, the results showed that glucose restriction led to a decrease in alpha-ketoglutarate (aKG), a key metabolite involved in histone demethylation. This resulted in decreased activity of aKG-dependent histone demethylases and increased histone hypermethylation, particularly on H3K27. The authors found that the polycomb repressor complex 2 (PRC2) component EZH2 plays a crucial role in this process by regulating the balance between histone demethylation and methylation.
Furthermore, glucose deprivation triggered the activation of the HIF1a signaling pathway, which is known to promote dedifferentiation and an aggressive phenotype. The authors discovered that EZH2 inhibits the prolyl-hydroxylase PHD3, a key regulator of HIF1a degradation under normoxic conditions. This inhibition led to HIF1a stabilization and subsequent activation of downstream targets like Slug, a key driver of EMT.
The authors demonstrated that targeting the EZH2/HIF1a/Slug axis with pharmacological inhibitors potentiated the antitumor effects of SGLT2 inhibitors, suggesting a potential strategy to overcome resistance to these drugs.
To validate the clinical relevance of this study, analysis of the TCGA database was conducted. It was revealed that the activation of the hypoxia pathway, as indicated by increased HIF1a expression, was associated with a worse prognosis in human LUAD patients. This suggested that the hypoxia pathway played a significant role in the clinical behavior of LUAD and could be a potential therapeutic target.
In conclusion, this study provided new insights into the relationship between glucose metabolism and cell differentiation in cancer. It revealed a previously unrecognized epigenetic adaptation mechanism by which cancer cells overcome the tumor-suppressive effects of glucose restriction. By inducing dedifferentiation and an aggressive phenotype through HIF1a activation, glucose deprivation may contribute to treatment resistance and poor clinical outcomes. The findings highlight the importance of considering the long-term effects of metabolic therapies and the need for combined approaches to prevent epigenetic adaptations and enhance treatment efficacy.
References:
Serio S, Pagiatakis C, Musolino E, Felicetta A, Carullo P, Laura Frances J, Papa L, Rozzi G, Salvarani N, Miragoli M, Gornati R, Bernardini G, Condorelli G, Papait R. Cardiac Aging Is Promoted by Pseudohypoxia Increasing p300-Induced Glycolysis. Circ Res. 2023 Sep 29;133(8):687-703.
Saggese P, Pandey A, Alcaraz M Jr, Fung E, Hall A, Yanagawa J, Rodriguez EF, Grogan TR, Giurato G, Nassa G, Salvati A, Shirihai OS, Weisz A, Dubinett SM, Scafoglio C. Glucose Deprivation Promotes Pseudohypoxia and Dedifferentiation in Lung Adenocarcinoma. Cancer Res. 2024 Jan 16;84(2):305-327.





Post comments