
CD36 plays a crucial role in the regulation of atherosclerosis progression and regression by facilitating macrophage lipid uptake. Recently, Zhang et al revealed that CD36 was a key player in the accelerated development of atherosclerosis in Card9-deficient mice. CARD9 is a signaling adaptor protein primarily expressed in macrophages and dendritic cells. It integrates signals from pattern recognition receptors like Toll-like receptors (TLRs), NOD2, and Dectin-1 to regulate innate immune responses. CARD9 has been found to be crucial for the production of interleukins such as IL-6, IL-17A, IFN-γ, and IL-22. It was observed that global deletion of Card9 accelerated atherosclerosis in Apoe-/- mice, highlighting the pro-atherogenic effects of Card9 deficiency. The results indicated that Card9 deficiency in Apoe-/- mice led to an inflammatory plaque phenotype with increased macrophage accumulation, necrotic core size, and collagen content. Moreover, Card9 deficiency dampened systemic pro-inflammatory cytokine signatures, altered T cell polarization, and promoted foam cell formation.
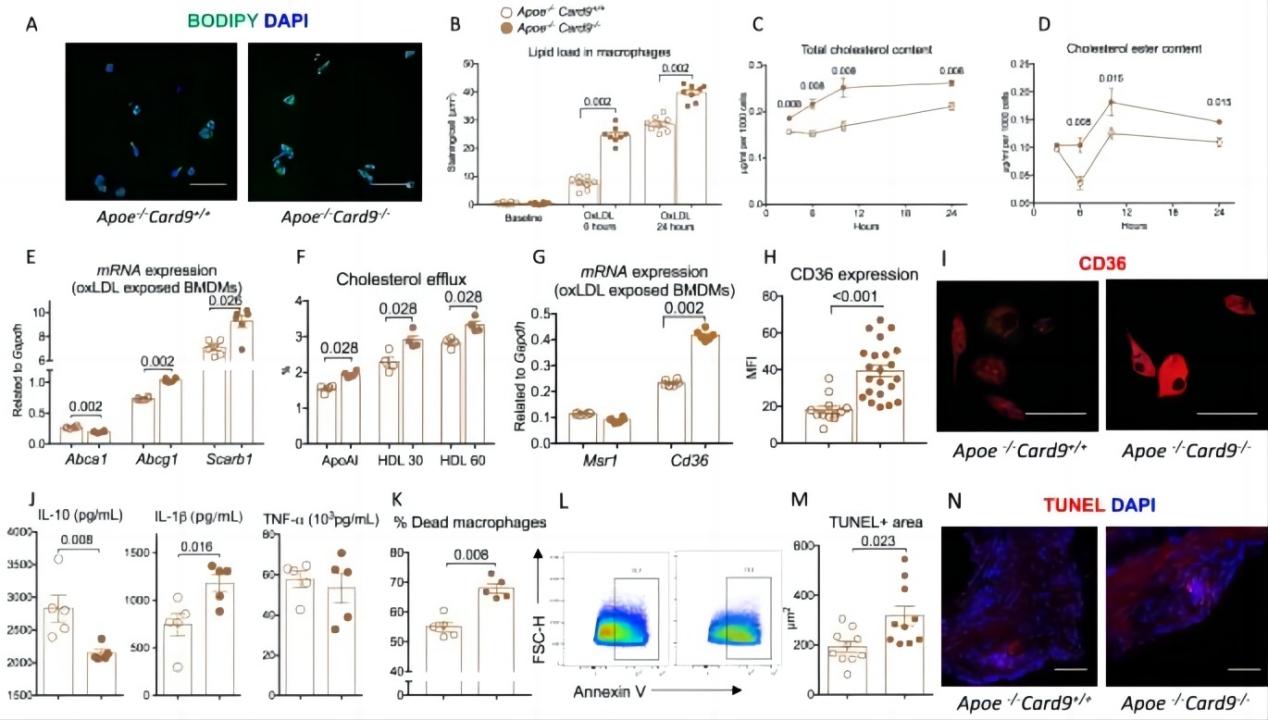
Fig.1:Card9 deficiency increased CD36 expression in macrophages and promotes lipid uptake and foam cell formation.
A Representative photomicrographs and quantitative analysis (B) of Bodipy+ foam cells after incubation of BMDMs from ApoeCard9 and ApoeCard9 mice with oxLDL during 6 and 24 h (2 pooled experiments, n = 8/group/timepoints), Scale bar 10 μm. C quantification of intracellular cholesterol content of BMDMs from ApoeCard9 and ApoeCard9 mice after exposure to ox-LDL (n = 6/group/timepoint). D Quantification of intracellular cholesterol ester of BMDMs from ApoeCard9 and ApoeCard9 mice after exposure to ox-LDL at 3, 6, 12, and 24 h (n = 6/group/timepoint). E Quantification of Abca1 and Abcg1 and Scarb1 mRNA expression in BMDMs from ApoeCard9 and ApoeCard9 mice after stimulation with oxLDL (n = 6/group). F Quantification of cholesterol efflux in presence of ApoAI or HDL (n = 4/group). G Quantification of Mrs1 and Cd36 mRNAs in BMDMs from ApoeCard9 and ApoeCard9 mice after stimulation with oxLDL (n = 6/group). H, I representative immunostaining and quantification of Cd36 expression by BMDMs from ApoeCard9 and ApoeCard9 mice after 24-h stimulation with ox-LDL (n = 12 ApoeCard9 and n = 22 ApoeCard9), Scale bar 10 μm. J cytokine production by BMDMs from ApoeCard9 and ApoeCard9 mice after stimulation (ELISA in the supernatant, n = 5/group) and oxLDL exposure. K, L Flow cytometry quantification of AnnexinV+ BMDMs from ApoeCard9 and ApoeCard9 mice after 24-h stimulation with oxLDL (100 μmol/l; n = 5/group). M, N representative photomicrographs and quantification of TUNEL+ cells in plaques from ApoeCard9 and ApoeCard9 mice (n = 10/group). Scale bar 20 μm. Data are presented as mean values ±SD. Two-tailed Mann–Whitney test. Source data are provided as a Source Data file.
Credit:DOI:https://doi.org/10.1038/s41467-023-40216-x
By using chimeric Ldlr-/- mice, where only hematopoietic cells (i.e., cells derived from the bone marrow) lack Card9, the study found that the pro-atherogenic effects might be specifically due to the absence of Card9 in myeloid cells, including macrophages. Additionally, it demonstrated that the effects of Card9 deficiency were independent of adaptive immunity, as evidenced by accelerated atherosclerosis in Apoe-/-Rag2-/-Card9-/- mice which lacked functional T, B, and NKT lymphocytes.
Furthermore, the study demonstrated that CD36 upregulation contributed to the acceleration of atherosclerosis in Card9-/- mice, and the pro-atherogenic effects of Card9 deficiency were abolished in Cd36-/-Card9-/- mice. These effects were mediated by the enhancement of CD36-dependent lipid uptake in macrophages from Card9-/- mice, which led to impaired autophagy. In vivo, pharmacological activation of autophagy using rapamycin and metformin abolished the pro-atherogenic effects of Card9 deficiency, which indicated that defective autophagy caused by Card9 deficiency might contribute to the subsequent lipid overload, cell death, and a pro-inflammatory phenotype.
In addition, the study demonstrated the clinical relevance of these findings by identifying changes in monocytes from CARD9-deficient patients. The monocytes from CARD9-deficient patients showed evidence of dysregulated immune responses, with increased production of pro-inflammatory cytokines and altered expression of genes involved in apoptosis, autophagy, atherosclerosis, NF-κB signaling, and TNFα signaling. Additionally, the detection of CARD9 in human atherosclerotic plaques added further weight to the translational relevance of the findings.
In summary, this comprehensive study established CARD9 as a key protective pathway in atherosclerosis, and the pro-atherogenic effects of Card9 deficiency were mediated by CD36-dependent defective autophagy. The findings provide a novel mechanistic insight into the role of CARD9 in atherosclerosis pathogenesis and have implications for the development of atherosclerosis-targeting therapies.
Another article published in Circulation Research found that Epsins also enhanced CD36-mediated lipid uptake by promoting CD36 endocytosis and recycling. The authors presented an insightful study on the role of endocytic adaptor proteins Epsin1 and Epsin2 in regulating cholesterol metabolism and transport in macrophages, and their impact on atherosclerosis progression and regression.
The authors employed single-cell RNA sequencing and a newly developed algorithm called MEBOCOST (Metabolite-mediated Cell Communication Modeling by Single Cell Transcriptome) to identify the role of Epsins in macrophage-mediated lipid metabolism regulation. They found that Epsins enhanced CD36-mediated lipid uptake by promoting CD36 endocytosis and recycling, while also inhibiting ABCG1-mediated cholesterol efflux. Specifically, Epsin1 interacts with CD36 via its Epsin N-terminal homology domain, which is critical for CD36-mediated lipid uptake and foam cell formation. Epsin-deficient macrophages showed increased cholesterol efflux, reversed cholesterol transport and decreased foam cell formation compared to wild-type macrophages.
To therapeutically target Epsins, the authors developed S2P-conjugated lipid nanoparticles encapsulating siRNAs targeting Epsin1/2. These nanoparticles effectively silenced Epsin expression in lesional macrophages and reduced plaque progression in early and advanced atherosclerosis mouse models. Furthermore, they promoted atheroma regression by diminishing inflammation and enhancing plaque stability.
In summary, this study provides evidence that Epsin inhibition in lesional macrophages has therapeutic benefits for treating advanced atherosclerosis by reducing lipid uptake and increasing cholesterol efflux. The innovative nanoparticle-mediated siRNA delivery approach represents a promising strategy for precision treatment of atherosclerosis. This research offers a comprehensive mechanistic understanding of the roles of Epsins in lipid metabolism and cholesterol transport, and it has significant potential implications for the development of new nanotherapeutic treatments for atherosclerosis.
Overall, these studies offer valuable insights into atherosclerosis pathogenesis, highlighting CARD9 and Epsin as potential therapeutic targets. These findings underscore the importance of CD36-mediated lipid uptake and foam cell formation in atherosclerosis and suggest new approaches for atherosclerosis prevention and treatment.
Zhang Y, Vandestienne M, Lavillegrand JR, Joffre J, Santos-Zas I, Lavelle A, Zhong X, Le Goff W, Guérin M, Al-Rifai R, Laurans L, Bruneval P, Guérin C, Diedisheim M, Migaud M, Puel A, Lanternier F, Casanova JL, Cochain C, Zernecke A, Saliba AE, Mokry M, Silvestre JS, Tedgui A, Mallat Z, Taleb S, Lenoir O, Vindis C, Camus SM, Sokol H, Ait-Oufella H. Genetic inhibition of CARD9 accelerates the development of atherosclerosis in mice through CD36 dependent-defective autophagy. Nat Commun. 2023 Aug 1;14(1):4622.
Cui K, Gao X, Wang B, Wu H, Arulsamy K, Dong Y, Xiao Y, Jiang X, Malovichko MV, Li K, Peng Q, Lu YW, Zhu B, Zheng R, Wong S, Cowan DB, Linton M, Srivastava S, Shi J, Chen K, Chen H. Epsin Nanotherapy Regulates Cholesterol Transport to Fortify Atheroma Regression. Circ Res. 2023 Jan 6;132(1):e22-e42.




Post comments