
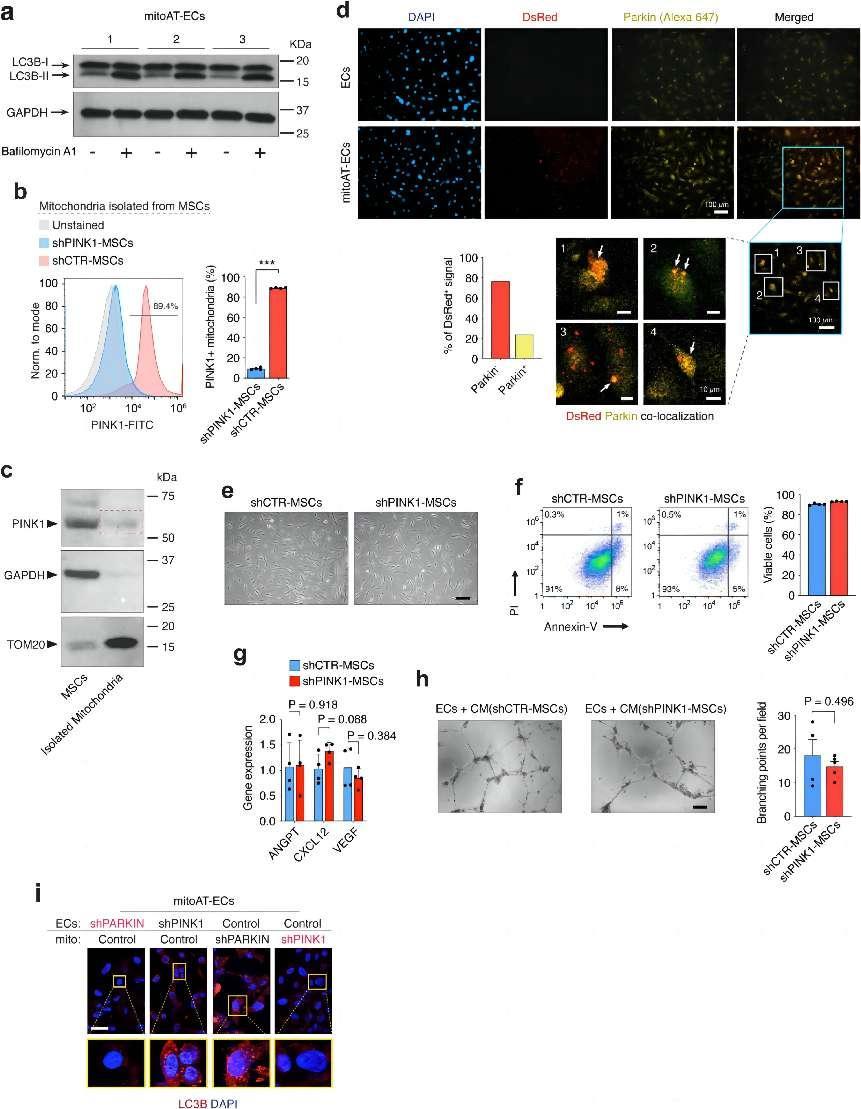
a, Analysis of autophagic flux in mitoAT-ECs, showing LC3B-I to LC3B-II conversion with Bafilomycin A1. b, Flow cytometry analysis showing PINK1 presence on isolated mitochondria from control MSCs (shCTR-MSCs) but not from shPINK1-MSCs. Quantification of the percentage of PINK1-positive in isolated mitochondria from control versus shPINK1-MSCs. ***P ≤ 0.001 (n = 4; unpaired two-tailed t-test). c, Western blot analysis demonstrating the presence of PINK1 (63 kDa) in lysates from both MSCs and isolated mitochondria (red line box), with TOM20 (16 kDa) as a mitochondrial marker and GAPDH (36 kDa) as a cytosolic control. d, Immunofluorescence indicates co-localization of exogenous DsRed+ mitochondria (Red) with endogenous Parkin (Alexa 647) in mitoAT-ECs 24 h post-transplantation (white arrows). Scale bar, 100 μm; insets #1–4, 10 µm. e–h, Evaluation of MSC viability and functionality after PINK1 silencing (shRNA). e, Morphological observations of MSCs with shRNA against PINK1 (shPINK1-MSCs) vs. control shRNA (shCTR-MSCs) show standard mesenchymal cell morphology. Scale bar, 100 μm. f, Flow cytometry using PI/Annexin-V highlights the high viability of shPINK1-MSCs after lentiviral transduction (n = 4). g, qPCR analysis measures angiogenic growth factor expression (ANGPT, CXCL12, VEGF) in shCTR-MSCs vs. shPINK1-MSCs (n = 4; unpaired two-tailed t-test). h, In vitro assay of EC vascular network formation, using conditioned medium (CM) from shPINK1-MSCs vs. shCTR-MSCs (n = 4; unpaired two-tailed t-test). Scale bar, 200 μm. i, Immunofluorescence detection of LC3B+ autophagosomes in mitoAT-ECs. Effects of Parkin and PINK1 silencing (shRNA) in either the donor MSC mitochondria (mito) or recipient ECs. DAPI denotes cell nuclei. Scale bar, 10 μm. All data are mean ± s.e.m. n are biological replicates (b,f,g,h). credit:https://doi.org/10.1038/s41586-024-07340-0
A groundbreaking study published in Nature has delved into the intricate world of cellular interactions, uncovering a novel mechanism by which mesenchymal stem cells (MSCs) aid in the successful engraftment of endothelial cells (ECs) in ischemic tissue. The research, employing a multifaceted approach, explored the role of mitochondrial transfer in enhancing EC survival and functionality. This discovery could have significant implications for the field of vascular cell therapy and the treatment of ischemic conditions.
In the first part of the study, the authors observed that the survival and engraftment of ECs was significantly enhanced when co-implanted with MSCs, but this was not the case when ECs were implanted alone. This led them to hypothesize that MSCs played a crucial role in promoting EC engraftment.
Next, they demonstrated that MSCs could transfer mitochondria to ECs through tunneling nanotubes (TNTs), a process that was crucial for successful EC engraftment. This was visualized using fluorescently labeled mitochondria in MSCs and flow cytometry to quantify the proportion of ECs receiving mitochondria.
The authors then sought to confirm that blocking mitochondrial transfer would impair EC engraftment. They achieved this by inhibiting TNT formation or mitochondrial mobilization in TNTs using gene silencing and anti-TNF antibody treatment. These methods significantly reduced EC engraftment.
To enhance EC engraftment without MSCs, the authors developed a novel strategy. They co-cultured ECs with MSCs to allow a subset of ECs to receive mitochondria through TNTs, resulting in enhanced engraftment capability.
Building upon these insights, the authors further demonstrated that artificial transplantation of exogenous mitochondria could enhance EC engraftment without the need for MSCs. This approach utilized isolated mitochondria from MSCs to transplant into ECs, which led to transient improvements in EC bioenergetics and engraftment ability.
Unexpectedly, the exogenous mitochondria did not need to be functional to induce these benefits. Depolarized or mtDNA-free mitochondria could also enhance EC function. Mechanistically, the exogenous mitochondria triggered mitophagy after internalization, leading to increased mitochondrial biogenesis.
The authors confirmed the involvement of mitophagy in mediating EC engraftment by inhibiting the PINK1-Parkin pathway, which was known to regulate mitophagy. This inhibition abrogated the enhanced engraftment ability of ECs with exogenous mitochondria.
In summary, this study revealed a mechanism by which MSCs facilitated EC engraftment through mitochondrial transfer, which in turn triggers mitophagy to enhance EC fitness. These findings have implications for vascular cell therapy, suggesting a potential approach for enhancing EC engraftment and revascularization of ischemic tissue. The authors' methods and conclusions contribute to a better understanding of how mitochondrial transfer impacts EC function and survival, paving the way for potential clinical applications.
Mitochondrial transfer can not only occur between mesenchymal cells and endothelial cells but also between many other types of cells, such as immune cells and tumor cells. Zhang et al. explored a novel phenomenon where T cells transferred mitochondria to cancer cells, thereby influencing their metabolic pathways, proliferation, and ultimate prognosis.
To investigate mitochondrial transfer, the authors conducted coculture experiments using cancer cells labeled with fluorescent mitochondrial markers and T cells isolated from mice. They then used scRNA-seq to analyze the single-cell transcriptomes of cocultured cancer cells, monocultured cancer cells, and T cells. The scRNA-seq data revealed that cocultured cancer cells expressed a mixture of T cell and cancer cell mitochondrial transcripts, indicating mitochondrial transfer.
To quantify mitochondrial transfer, the authors developed MERCI (mitochondrial-enabled reconstruction of cellular interactions), a statistical deconvolution method that used the expression of mitochondrial genes and single nucleotide variants (SNVs) to predict mitochondrial receiver cells and their relative mitochondrial compositions. MERCI was validated and benchmarked using experimental ground-truth data and was found to accurately predict the recipient cells and their mitochondrial compositions.
The authors applied MERCI to human cancer samples and identified a distinct mitochondrial transfer phenotype in cancer cells. This phenotype was characterized by upregulated genes in pathways related to energy production, cytoskeleton remodeling, and TNF-α signaling. Analysis of large patient cohorts revealed that the mitochondrial transfer phenotype was associated with increased cancer cell proliferation and poor prognosis across different cancer types.
The study identified 17 gene signatures associated with mitochondrial transfer, which served as biomarkers for predicting mitochondrial transfer in cancer cells. The TMT (tumor mitochondrial transfer) score, calculated from these gene signatures, was found to be associated with increased proliferation and poor prognosis in cancer patients.
In summary, this study uncovered a previously unreported mitochondrial transfer phenomenon from T cells to cancer cells, provided a computational method to trace this transfer, and demonstrated the functional impact of mitochondrial transfer on cancer cell metabolism, proliferation, and prognosis. The identified gene signatures and TMT score may serve as potential biomarkers and therapeutic targets for mitochondrial transfer in cancer. Overall, these research presented in this article underscored the critical role of cellular communication and mitochondrial dynamics in maintaining tissue health and fighting diseases such as ischemia and cancer.
Lin RZ, Im GB, Luo AC, Zhu Y, Hong X, Neumeyer J, Tang HW, Perrimon N, Melero-Martin JM. Mitochondrial transfer mediates endothelial cell engraftment through mitophagy. Nature. 2024 May;629(8012):660-668.
Zhang H, Yu X, Ye J, Li H, Hu J, Tan Y, Fang Y, Akbay E, Yu F, Weng C, Sankaran VG, Bachoo RM, Maher E, Minna J, Zhang A, Li B. Systematic investigation of mitochondrial transfer between cancer cells and T cells at single-cell resolution. Cancer Cell. 2023 Oct 9;41(10):1788-1802.e10.




Post comments