
Credit:A tryptophan-derived uremic metabolite/Ahr/Pdk4 axis governs skeletal muscle mitochondrial energetics in chronic kidney disease.
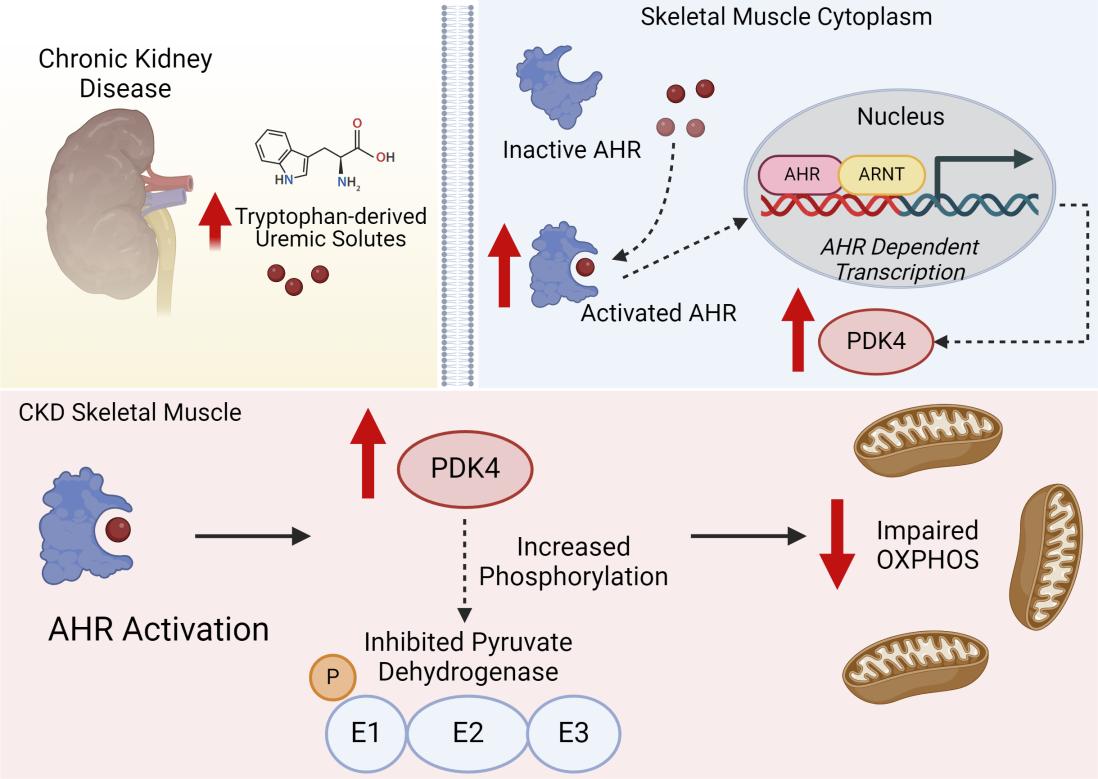
Chronic kidney disease (CKD) is a prevalent condition that leads to significant muscle dysfunction, characterized by reduced muscle mass, strength, and exercise intolerance. While the mechanisms driving muscle wasting in CKD are well-documented, less is understood about the metabolic deficiencies observed in skeletal muscle. This comprehensive study conducted by Thome et. al. delved into the complex interplay between chronic kidney disease (CKD), uremic metabolites, and skeletal muscle dysfunction, with a particular emphasis on the aryl hydrocarbon receptor (AHR) as a key mediator. The authors employed a multifaceted approach, utilizing both human and animal models, to unravel the intricate mechanisms underlying CKD-related muscle wasting and mitochondrial dysfunction.
The research began with a thorough examination of skeletal muscle biopsies from CKD patients and healthy controls. Utilizing quantitative PCR (qPCR) and immunoblotting techniques, the authors observed a significant upregulation of AHR and its downstream target genes (CYP1A1 and CYP1B1) in CKD muscle, indicating AHR activation. Moreover, they established a strong inverse correlation between the expression of CYP1A1 and mitochondrial respiration rates, suggesting a direct link between AHR activation and mitochondrial dysfunction. These findings laid the foundation for the subsequent animal studies, providing evidence of AHR activation in human CKD muscle and its association with impaired mitochondrial function.
The authors employed a well-established mouse model of CKD, utilizing adenine supplementation to induce kidney damage and elevate uremic metabolite levels. To further augment uremia and AHR activation, they administered probenecid, an organic anion transporter inhibitor, to a subset of CKD mice. This experimental design allowed for a nuanced investigation of the effects of varying degrees of uremia on AHR activation and mitochondrial function.
To assess the impact of AHR activation on muscle mitochondrial function, the authors generated a muscle-specific AHR knockout mouse model (AHRmKO) using the Cre-loxP system. They confirmed successful deletion of AHR in skeletal muscle and demonstrated that AHRmKO mice exhibited significantly improved mitochondrial OXPHOS function, particularly in male mice and when mitochondria were fueled by carbohydrates. This improvement was abolished when AHR was deleted in the context of high uremic metabolite levels, highlighting the critical role of AHR activation in mediating the effects of uremic toxins on mitochondrial function.
Intriguingly, the authors observed sex-specific differences in the response to AHR deletion. While male AHRmKO mice experienced significant improvement in mitochondrial function, female AHRmKO mice did not show similar benefits. This suggested potential differences in AHR biology or muscle mitochondrial function between the sexes, warranting further investigation to understand the underlying mechanisms and their implications for CKD management.
To elucidate the mechanisms underlying AHR-mediated mitochondrial dysfunction, the authors focused on the pyruvate dehydrogenase (PDH) complex, a crucial regulator of pyruvate metabolism. They found that AHR activation led to increased expression of pyruvate dehydrogenase kinase 4 (PDK4), a negative regulator of PDH activity. This, in turn, resulted in phosphorylation and inactivation of the PDH enzyme, impairing the ability of mitochondria to utilize carbohydrates as a fuel source. This impaired carbohydrate oxidation was thought to contribute to the mitochondrial dysfunction observed in CKD muscle.
To further validate the role of AHR activation in mitochondrial dysfunction, the authors expressed a constitutively active form of AHR (CAAHR) in the skeletal muscle of healthy mice using adeno-associated virus (AAV) technology. They observed a significant decrease in mitochondrial OXPHOS function in CAAHR-expressing mice, confirming the detrimental effects of AHR activation on muscle metabolism.
To determine whether AHR directly regulated PDK4 expression, the authors utilized transposase-accessible chromatin sequencing (ATAC-Seq) to analyze chromatin accessibility in CAAHR-expressing muscle. They identified significant differences in chromatin accessibility around the PDK4 promoter region between CAAHR and control muscle, suggesting direct transcriptional regulation of PDK4 by AHR. Furthermore, they demonstrated that a transcriptionally inactive mutant of CAAHR (R39D) failed to induce PDK4 expression or impair mitochondrial function, further confirming the requirement for AHR transcriptional activity in mediating these effects.
The study provided compelling evidence for the role of AHR activation in mediating CKD-related muscle dysfunction and mitochondrial dysfunction. The findings suggested that the accumulation of tryptophan-derived uremic metabolites, such as kynurenines and indoles, triggered AHR activation in skeletal muscle, leading to increased PDK4 expression and subsequent impairment of carbohydrate oxidation. This impaired mitochondrial function contributed to muscle wasting and reduced exercise tolerance observed in CKD patients.
While the study provides valuable insights into the role of AHR in CKD-related muscle dysfunction, it is important to acknowledge potential limitations. The study primarily focuses on mitochondrial function and does not address other aspects of muscle dysfunction, such as muscle wasting and contractile dysfunction. Additionally, the study utilizes a specific mouse model of CKD and the findings may not be directly applicable to all forms of CKD. Further research is needed to investigate the role of AHR in other aspects of muscle dysfunction and to determine the relevance of these findings to human CKD. Additionally, the sex-specific differences in the response to AHR deletion are intriguing and warrant further investigation. Potential mechanisms for these differences could include variations in AHR expression, sensitivity, or downstream signaling pathways between the sexes. Understanding these sex-specific effects is crucial for developing targeted therapies for CKD-related muscle dysfunction.
reference:
Thome T, Vugman NA, Stone LE, Wimberly K, Scali ST, Ryan TE. A tryptophan-derived uremic metabolite/Ahr/Pdk4 axis governs skeletal muscle mitochondrial energetics in chronic kidney disease. JCI Insight. 2024 Apr 23;9(10):e178372.






Post comments