
‘The behaviour of all living organisms can be understood by the fluttering and wiggling of atoms.’ As Nobel Prize-winning physicist Richard Feynman famously said, the essence of the biological world is the never-ending process of atomic motion. Exploring the movement of biomolecules and the interactions between molecules is crucial to deciphering the mechanisms behind life's activities, as well as to designing and discovering new drugs, vaccines, and biomaterials.
In recent years, with the development of deep learning techniques and the rapid increase in GPU computational power, artificial intelligence has played an increasingly important role in the field of protein research. 2024's Nobel Prize in Chemistry was awarded to research on protein structure prediction and protein design. While computationally predicting static protein crystal structures is close to or at the same level of accuracy as biological experiments, how to use AI to accurately characterise the dynamically changing behaviour of proteins at the atomic level is a challenge that still needs to be solved and is much more difficult.
Recently, Microsoft Research AI for Science researcher Tong Wang and his team have made significant progress in AI-driven molecular dynamics simulation research over a four-year period, and the results have been published online in the world's top scientific journal Nature in the form of a long article. The results have been published online in the form of a long article in the main issue of the world's top scientific journal Nature.

AI-driven molecular dynamics simulations
Molecular Dynamics (MD) is a technical tool for simulating the motion of molecules and atoms in real biological cells. Dynamics simulation is generally 1 femtosecond (10-15 seconds) as a step simulation, through hundreds of millions to hundreds of billions of steps simulation, reflecting the spatial and temporal movement of protein molecules in the cell. After more than half a century of development, molecular dynamics simulation can be divided into two categories: classical MD Simulation and quantum simulation.
Classical simulation uses Newtonian mechanics as a force field to drive the motion of atoms and molecules, which has the characteristics of fast speed and wide applicability. For more than half a century, classical simulation has been widely used in the study of the dynamics of biological macromolecules such as proteins, and was awarded the Nobel Prize in Chemistry in 2013. However, classical simulations using Newtonian force fields lack the accuracy of the force fields and are unable to simulate the behaviour of molecular bonding and bond breaking and other electron migration, which stretches the limits of high-precision free energy calculations, virtual screening of drugs, biochemical reactions, and so on.
In contrast to classical simulation, the quantum simulation method represented by Density Functional Theory (DFT) adopts quantum mechanical force field, and the motion description of atoms can reach the accuracy of ab initio calculation. With its complete theoretical foundation and wide application in the field of computational chemistry, Density Functional Theory was awarded the Nobel Prize in 1998. However, due to the extremely high computational cost, quantum simulation can neither be directly applied to the study of biological macromolecules such as proteins, nor be simulated for a long time.
How to break the technical bottleneck between classical and quantum simulation and achieve all-atom simulation of biological macromolecules such as proteins with quantum-level accuracy has been a major challenge in the field for more than half a century.
In order to address this major challenge, researchers at the Centre for Scientific Intelligence at Microsoft Research have designed AI2BMD (AI powered ab initio biomolecular dynamics), an AI-based molecular dynamics simulation system. The system efficiently performs all-atom simulations of various types of proteins with ab initio precision (i.e., quantum-level accuracy). This innovation achieves a trade-off in biomolecular simulation that was previously unattainable with standard simulation techniques - higher accuracy than classical simulation, and computational speeds that are orders of magnitude ahead of DFT and other quantum mechanical methods, albeit at a higher computational cost than classical simulation. AI2BMD holds the promise of unlocking many new capabilities in biomolecular modelling, particularly AI2BMD is expected to unlock new capabilities in biomolecular modelling, especially in research processes such as protein-drug interactions, where high computational accuracy is required.
Animated demonstration of AI-driven molecular dynamics simulation
In-depth AI2BMD technology innovation
One of the most important components of a molecular dynamics simulation is the construction of a force field. At each step of the simulation, the force field calculates the energy of the molecule and the force applied to each atom, which drives the motion of the entire molecule. Classical simulations use Newtonian force fields and quantum simulations use quantum mechanical force fields. To build AI-driven molecular dynamics simulations, the biggest challenge is the generalisability of deep learning models, i.e., the accuracy of models trained on known molecules in predicting the energy and force of various types of unknown protein molecules. To this end, the research team designed a protein fragment-based, generalisable segmentation technique to segment various types of protein molecules into 21 generic protein fragments. The construction of the dataset and the training of the model are all based on the generic protein fragments, thus achieving a generic solution for all types of protein molecules (Figure 1).
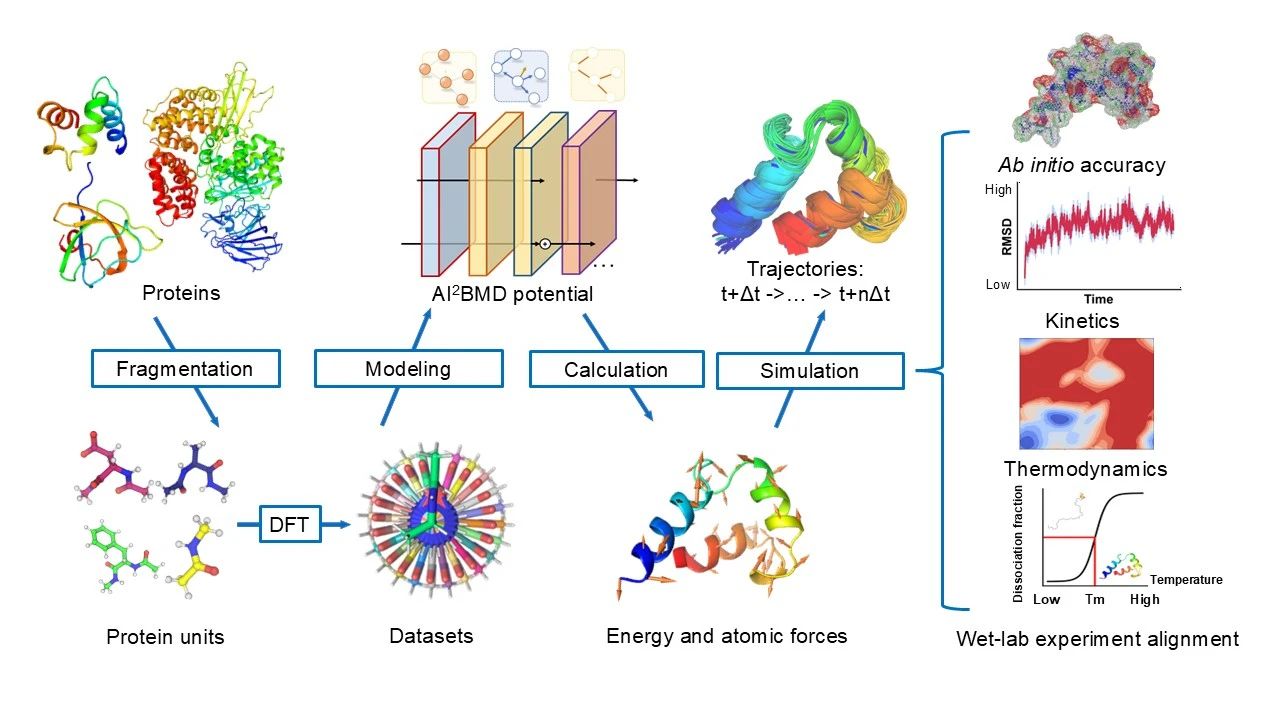
Figure 1: Flow chart of AI2BMD technology
Based on the universal protein segmentation scheme, the research team further constructed the Protein Unit Dataset (https://github.com/microsoft/AI2BMD), which contains more than twenty million pieces of data and is currently the world's largest protein fragment dataset with quantum-level accuracy. The researchers selected ViSNet, a previously developed network model for generic molecular geometry modelling, and trained it on Protein Unit Dataset to serve as a force field for AI2BMD. Considering the efficiency of molecular simulation, the team proposed a new client-server architecture, which can compress each simulation step to the order of tens of milliseconds by dynamically scheduling CPUs and GPUs. The researchers used AI2BMD to analyse the kinetics and thermodynamics of various proteins, and the results showed better results than classical simulations in various aspects such as protein folding free energy calculation and conformational space exploration.
Technological innovations in biomolecular modelling
AI2BMD demonstrates an innovative departure from previous classical simulations of protein molecules in the following ways:
Quantum-level accuracy: AI2BMD enables all-atom protein dynamics simulations with quantum-level accuracy through a generalisable ‘machine-learning force field’, a model of the interactions between atoms and molecules constructed through machine-learning models.
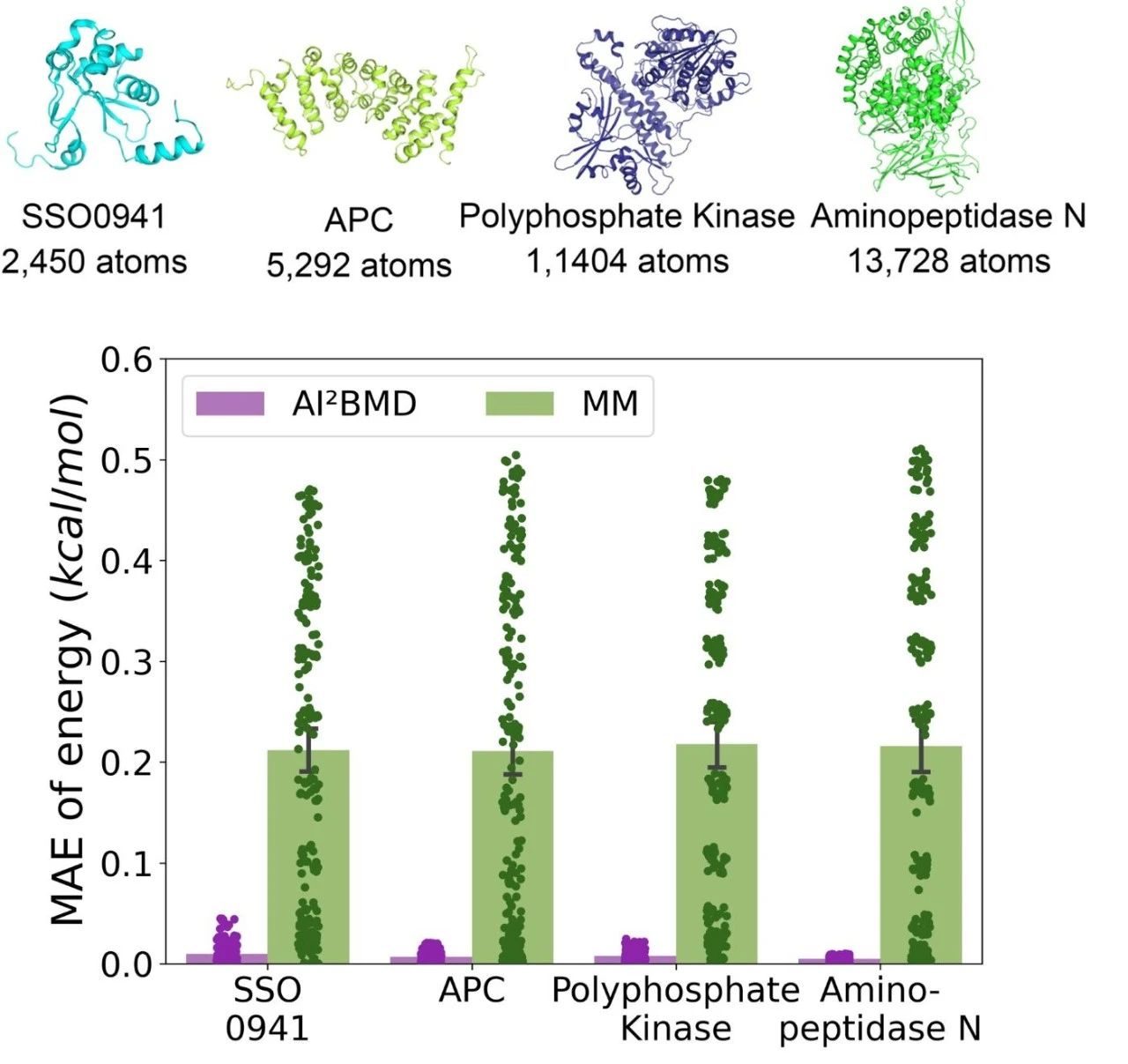
Figure 2: Comparison of errors between AI2BMD and classical kinetic simulations for different protein energy calculations
Generalisability: AI2BMD addresses for the first time the challenge of generalising machine learning force fields to simulate protein dynamics, demonstrating robustness to all-atom simulations of a wide range of proteins.
All-atom simulation compatibility: In contrast to hybrid simulation techniques combining quantum and classical simulations, AI2BMD extends quantum-level precision calculations to the entire protein molecule and does not require any a priori knowledge about the protein. This removes potential incompatibilities between quantum and classical simulation calculations of proteins and increases the computational speed in the quantum simulation region by several orders of magnitude, bringing near ab initio calculations of all-atom proteins closer to reality. As a result, AI2BMD paves the way for many downstream applications and provides new perspectives for characterising complex biomolecular dynamics.
Efficiency: AI2BMD is orders of magnitude faster than DFT and other quantum simulations.AI2BMD supports quantum-level accuracy calculations for proteins with more than 10,000 atoms, making it one of the fastest AI-driven molecular dynamics simulation programs in many subject areas.
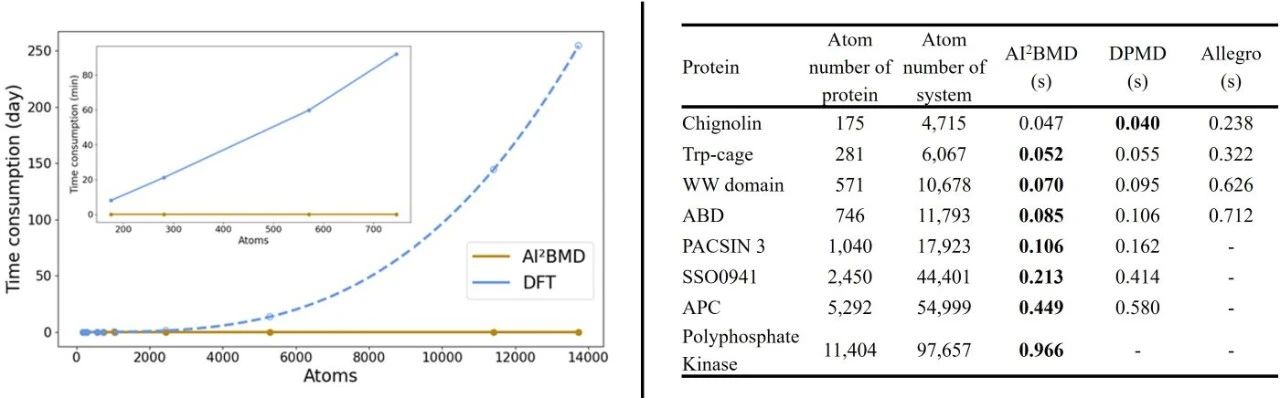
Figure 3: Comparison of the speed of AI2BMD with DFT and other AI-driven dynamics simulation software
Multiplicity of conformational exploration: unlike classical simulations, AI2BMD does not impose any constraints on bond lengths, bond angles, dihedral angles, and so on. As shown in Fig. 4, during the simulation of protein folding and defolding with AI2BMD and classical simulation, respectively, AI2BMD explores more possible conformational spaces that cannot be detected by classical simulation. Therefore, AI2BMD provides more opportunities and possibilities to study the flexible movement of proteins, enzyme catalysis, metastable regulation, intrinsically disordered proteins, etc. during drug target binding.
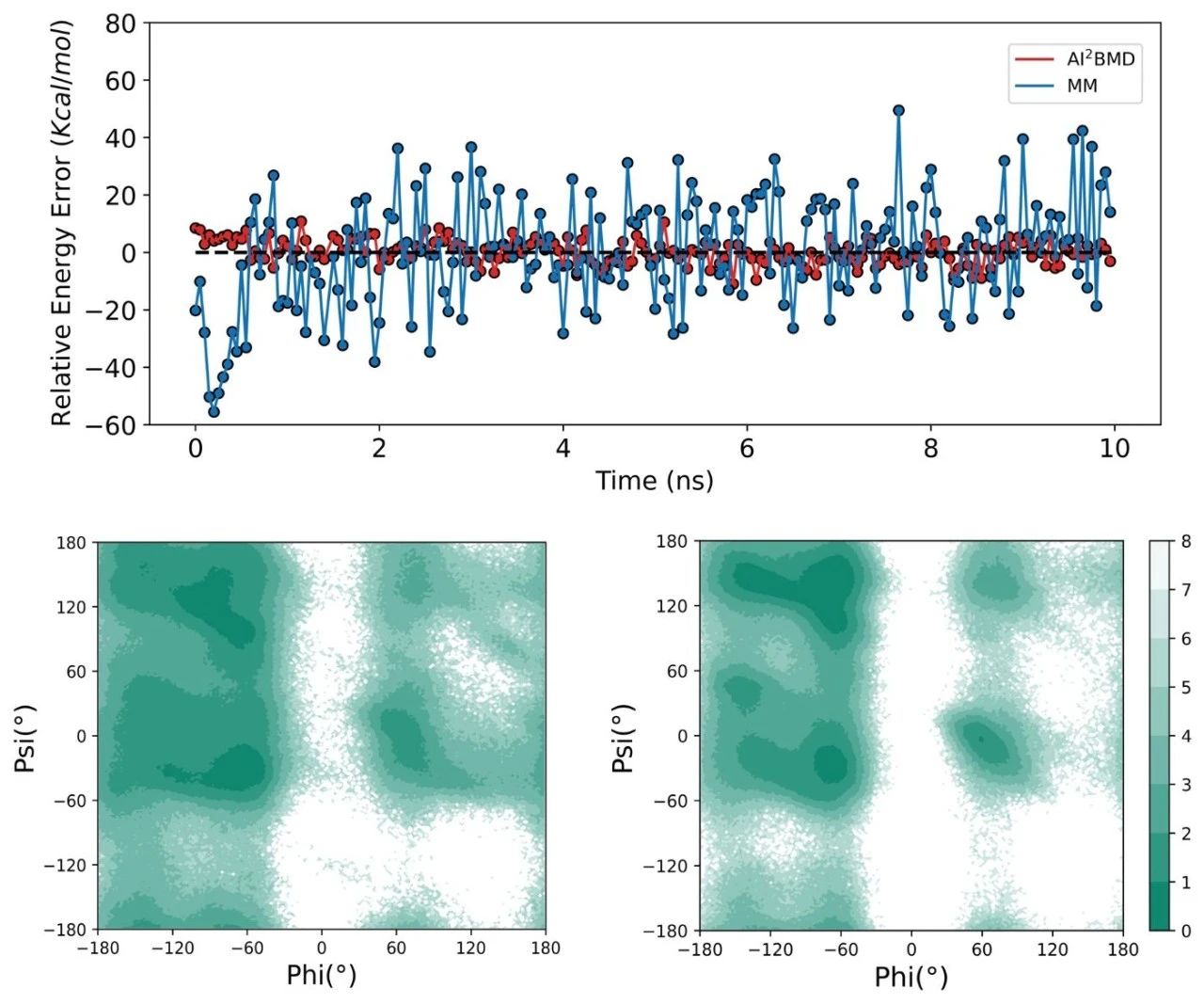
Figure 4: Simulation performance of AI2BMD versus classical simulation in protein Chignolin folding process
Consistency with biological experiments: Compared with classical and hybrid simulations, AI2BMD shows higher consistency with biological experiments in terms of J-coupling, enthalpy change, heat capacity, free energy of folding, melting temperature and pKa.
Applications and Perspectives
Achieving quantum-level accuracy in biomolecular simulations is challenging, but it holds great potential for unravelling the mysteries of biological systems and designing novel biomaterials and drugs. This breakthrough is a testament to AI for Science's vision of harnessing the power of artificial intelligence to revolutionise scientific discovery. AI2BMD achieves a balance of accuracy, stability and generalisation of the machine learning force field for molecular dynamics simulation applications, improving the accuracy of calculations of energy and atomic stresses while enabling more accurate calculations and estimations of various properties of proteins.
A key application scenario for AI2BMD is high-precision binding energy calculation between target proteins and drug molecules in drug discovery. In the first global AI drug discovery competition in 2023, AI2BMD and its AI powerhouse, ViSNet, accurately identified potential drug molecules that bind to multiple targets of the neocoronavirus, achieving the best prediction in all tasks and winning the championship.
In 2022, Microsoft Research also partnered with the Global Health Drug Discovery Institute (GHDDI), a non-profit organisation established by the Gates Foundation, the Beijing Municipal Government, and Tsinghua University, to apply AI technology to drug design. GHDDI is a non-profit organisation established by the Gates Foundation, the Beijing Municipal Government and Tsinghua University to develop drugs to treat diseases such as tuberculosis and malaria, which disproportionately affect low- and middle-income countries (LMIC). Microsoft Research is working closely with GHDDI to accelerate the drug discovery process through AI2BMD and other artificial intelligence technologies.
AI2BMD not only advances research on scientific questions, but also facilitates new biomedical research in areas such as drug discovery, protein design and enzyme engineering. Accurate and efficient characterisation of protein dynamics using AI2BMD is driving the development of scientific and technological innovations and stimulating a wide range of interest in the exploration of biological mechanisms in the scientific community.





Post comments