
Credit:2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis.
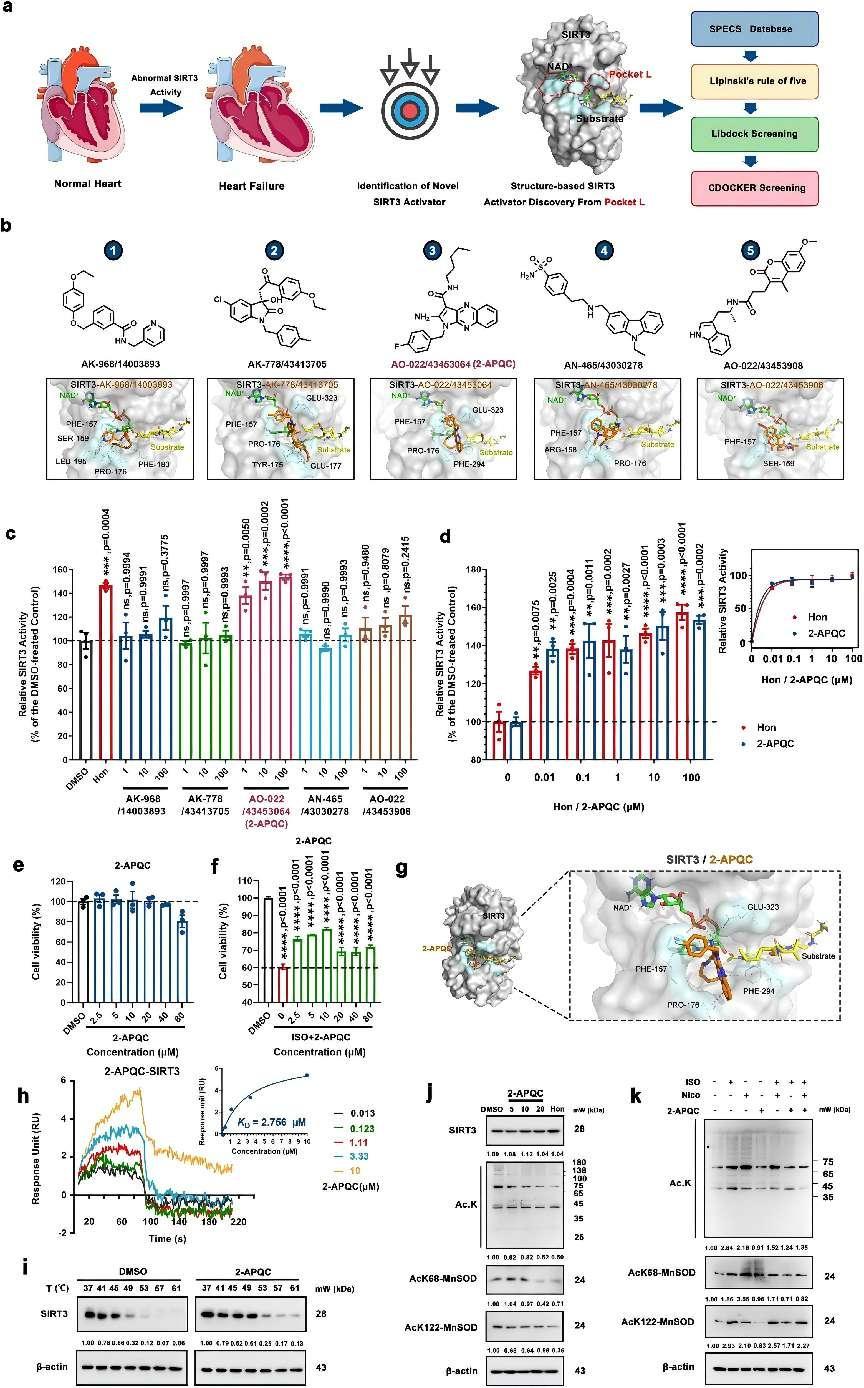
Structure-based screening of small-molecule activators of SIRT3 and identifies 2-APQC activates SIRT3 deacetylase activity in H9c2 cells. a The structure of SIRT3 and virtual screening scheme model for the discovery of SIRT3 activator. Created with biorender.com and https://smart.servier.com. b The 5 candidate compounds based on virtual screening and molecular docking. c The relative SIRT3 deacetylation activity after the candidate compounds (1, 10, 100 μM) and Honokiol (Hon) (10 μM) treatment. Data are present as mean ± s.e.m, n = 3. d The relative SIRT3 deacetylation activity after Hon or 2-APQC (0.01, 0.1 1, 10, 100 μM) treatment. Data are present as mean ± s.e.m, n = 3. e H9c2 cells were treated with indicated concentration of 2-APQC for 24 h, and cell viability was measured by the MTT assay. Data are present as mean ± s.e.m, n = 3. f H9c2 cells were treated with indicated concentration of 2-APQC for 24 h, and then treated with isoproterenol (ISO) for 48 h, the cell viability was measured by the MTT assay. Data are present as mean ± s.e.m, n = 3. g The molecular docking sketch map to indicate the binding mode between SIRT3 and 2-APQC. h Surface plasmon resonance assay detected the Kd value of SIRT3 in H9c2 cells treated with 2-APQC. i Cellular thermal shift assay detected the thermal stability of SIRT3 in H9c2 cells treated with 2-APQC. j Western blot analysis of SIRT3, acetylated-lysine, acetylated manganese superoxide dismutase 2 (MnSOD2) at K68 and K122 protein expression in H9c2 cells treated with 2-APQC for 24 h. k Western blot analysis of acetylated-lysine, acetylated MnSOD2 at K68 and K122 protein expression in H9c2 cells treated with 2-APQC for 24 h, together with or without ISO/nicotinamide (Nico) as the research design. β-actin, loading control. ns no significance; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Hon Honokiol, Nico nicotinamide, ISO isoproterenol
Heart failure (HF) is a multifaceted clinical syndrome characterized by impaired heart function, leading to reduced cardiac output and compromised organ perfusion. The pathophysiology of HF is complex, involving a constellation of molecular and cellular events that ultimately result in structural and functional alterations within the heart. Among these, myocardial hypertrophy and fibrosis are particularly significant contributors to the progression of HF, leading to reduced cardiac compliance, increased afterload, and ultimately, heart failure. Therefore, identifying effective strategies to target and modulate these processes holds immense potential for the treatment of HF.
Mitochondria, the energy powerhouses of the cell, play a crucial role in maintaining cellular homeostasis and are intimately involved in the pathophysiology of HF. Mitochondrial dysfunction, characterized by impaired energy production, increased reactive oxygen species (ROS) production, and altered redox balance, is a hallmark of HF. This dysfunction not only contributes to the development of myocardial hypertrophy and fibrosis but also exacerbates existing cardiac damage, leading to a vicious cycle of worsening heart failure. Therefore, targeting mitochondrial dysfunction and restoring mitochondrial homeostasis represents a promising therapeutic strategy for HF.
Sirtuin 3 (SIRT3), a member of the sirtuin family of proteins, is a key regulator of mitochondrial function and is emerging as a pivotal player in the pathophysiology of HF. SIRT3 functions as a NAD+-dependent deacetylase, modulating the acetylation status of numerous mitochondrial proteins, including those involved in oxidative stress, energy metabolism, and mitochondrial biogenesis. Emerging evidence suggests that SIRT3 plays a crucial role in the development of myocardial hypertrophy and fibrosis, primarily through its deacetylation modifications. For instance, SIRT3 can deacetylate glycogen synthase kinase 3 beta (GSK3beta) at Lys15, thereby inhibiting its activity and preventing excessive cell growth and proliferation, which is a key event in the development of myocardial hypertrophy. Additionally, SIRT3 can inhibit the TGF-beta/Smad3-mediated expression of fibrosis-related proteins, such as collagen and fibronectin, thereby reducing collagen deposition and fibrosis. Furthermore, SIRT3 can also inhibit the differentiation of cardiac fibroblasts into myofibroblasts, which are the primary cell type responsible for the synthesis and secretion of extracellular matrix proteins in the heart. These actions collectively suggest that SIRT3 plays a crucial role in maintaining myocardial homeostasis and preventing the development of HF.
While the potential of SIRT3 as a therapeutic target for HF is promising, the development of selective and effective SIRT3 activators remains a significant challenge. Several small-molecule compounds have been identified that can activate SIRT3 or upregulate its expression, such as honokiol, licorice isoflavone A, LCZ696, Oroxylin A, and resveratrol. However, the selectivity and efficacy of these compounds in targeting SIRT3 and improving HF remain limited. Therefore, the identification of novel, targeted SIRT3 activators with improved therapeutic potential is crucial for the development of effective HF therapies.
Building upon this knowledge, a recent study published in Signal Transduction and Targeted Therapy explored the potential of SIRT3 activators in alleviating myocardial hypertrophy and fibrosis, two key features of HF. The researchers employed structure-based drug design to identify a novel small-molecule activator of SIRT3, named 2-APQC. Through in vitro and in vivo experiments, they demonstrated that 2-APQC effectively alleviated isoproterenol (ISO)-induced cardiac hypertrophy and myocardial fibrosis in rat models. Importantly, the protective effects of 2-APQC were abrogated in SIRT3 knockout mice, confirming its dependency on SIRT3 for its beneficial effects.
The study further investigated the molecular mechanisms underlying the cardioprotective effects of 2-APQC. They found that 2-APQC inhibited the mammalian target of rapamycin (mTOR)-p70 ribosomal protein S6 kinase (p70S6K), c-jun N-terminal kinase (JNK), and transforming growth factor-β (TGF-β)/small mother against decapentaplegic 3 (Smad3) pathways, thereby improving ISO-induced cardiac hypertrophy and myocardial fibrosis. RNA-seq analyses revealed a close association between the SIRT3-pyrroline-5-carboxylate reductase 1 (PYCR1) axis and HF. By activating PYCR1, 2-APQC enhanced mitochondrial proline metabolism, inhibited reactive oxygen species (ROS)-p38 mitogen activated protein kinase (p38MAPK) pathway, and protected against ISO-induced mitochondrial oxidative damage. Moreover, activation of SIRT3 by 2-APQC facilitated AMP-activated protein kinase (AMPK)-Parkin axis to inhibit ISO-induced necrosis.
In conclusion, the study presents compelling evidence that 2-APQC is a novel, targeted SIRT3 activator with significant potential for the treatment of HF. By modulating mitochondrial homeostasis, 2-APQC could provide a promising therapeutic strategy for the management of HF, offering hope for the millions of individuals affected by this devastating disease.
Reference:
Peng F, Liao M, Jin W, Liu W, Li Z, Fan Z, Zou L, Chen S, Zhu L, Zhao Q, Zhan G, Ouyang L, Peng C, Han B, Zhang J, Fu L. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal Transduct Target Ther. 2024 May 15;9(1):133.






Post comments