
FEBRUARY 29, 2024
by The Hospital for Sick Children
Using a novel methodology, researchers at The Hospital for Sick Children (SickKids) are the first in pediatric research to use data from an international real-world cohort to overcome the barriers associated with conducting randomized clinical trials in children with rare diseases.
The gold standard for evaluating new therapeutics is through randomized clinical trials, where one group of individuals receives treatment while another does not. Unfortunately, conducting this type of clinical trial proves challenging for many rare conditions due to the limited number of individuals with the condition, making meaningful comparisons difficult. Additionally, in pediatrics, it can be unethical to give a potential treatment to some children and not others.
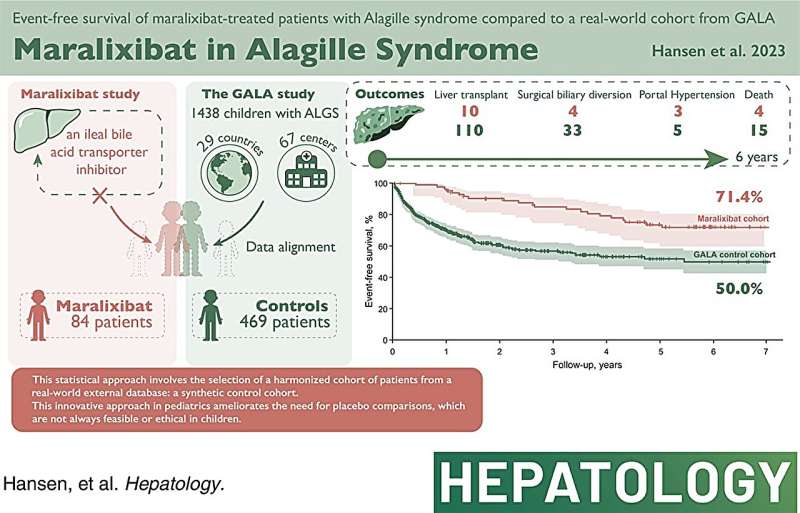
In a study published in Hepatology, a team of SickKids scientists developed an innovative and robust statistical approach which may eliminate the need for traditional randomized clinical trials, demonstrating the effectiveness of a medication in reducing disease progression and liver transplants in children with Alagille syndrome (ALGS).
"By using a global database, we were able to mimic high-quality clinical trial results by comparing present-day outcomes to historical data," says Dr. Binita Kamath, Senior Associate Scientist in the Developmental & Stem Cell Biology program and Principal Investigator and senior author of the study.
"
Not only do our findings show a marked improvement in liver-related outcomes for children with ALGS treated with this medication, but they also present a path forward for randomized clinical trials for other rare diseases."
Existing medication reduces risk of liver-related outcomes in children with ALGS
ALGS is a rare genetic disorder in which a patient has an inadequate number of bile ducts, which usually drain bile from the liver into the intestine. The bile then builds up and causes liver damage. Between 60 to 75 percent of patients with ALGS undergo a liver transplant before they reach adulthood.
Due to the rarity and severity of the condition, clinical trials have historically been difficult to conduct, especially over longer durations of time, since it is unethical and not feasible to have children take a placebo for several years.
In a previous study describing an international cohort of children with ALGS, Kamath and Shannon Vandriel established an international database of clinical, genetic and laboratory data in children and young adults with ALGS called The Global ALagille Alliance (GALA) study.
In this study, using the data of 469 untreated patients from GALA, the research team were able to compare their long-term outcomes to 84 children being treated with maralixibat, a medication approved in the US and Europe and recently in Canada for symptomatic relief of the severe itching experienced by patients with ALGS.
Using this innovative approach, the research team found that over six years, children taking maralixibat showed a 70 percent improvement in event-free survival and a 67 percent improvement in transplant-free survival.
"While the current indication for maralixibat is for itching, our data showed that over a longer period, the medication actually reduces the rate of liver transplants in patients with ALGS," says Kamath, who is also a Staff Physician in the Division of Gastroenterology, Hepatology and Nutrition.
Laying the groundwork for use in other clinical trials
In addition to these benefits, the methodology used by the research team has ground-breaking implications for the development and implementation of clinical trials for patients with rare diseases.
Kamath and Vandriel believe similar approaches could be applied to any rare disease with enough historical data.
"Our research provides an alternative to the challenges associated with recruiting patients with life-threatening conditions or debilitating symptoms for long-term clinical trials," says Kamath. "While our study was specific to patients with ALGS, I hope this sparks a renewed interest in international databases of individuals with rare diseases which can provide real-world data that can be used to help evaluate new therapies."
More information: Bettina E. Hansen et al, Event-free survival of maralixibat-treated patients with Alagille syndrome compared to a real-world cohort from GALA, Hepatology (2023). DOI: 10.1097/HEP.0000000000000727
Journal information: Hepatology
Provided by The Hospital for Sick Children





Post comments