
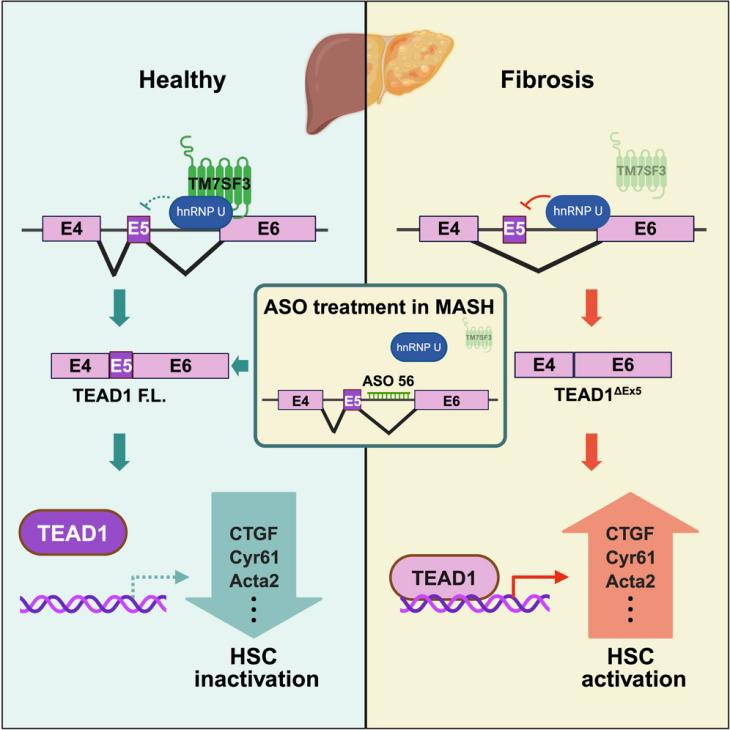
Credit:TM7SF3 controls TEAD1 splicing to prevent MASH-induced liver fibrosis.
Approximately 25% of patients with metabolic dysfunction-associated steatotic liver disease (MASLD) progress to metabolic dysfunction-associated steatohepatitis (MASH). MASH is characterized by hepatic steatosis, inflammation, hepatocyte injury (such as ballooning degeneration), and fibrosis. This condition can further progress to cirrhosis, hepatocellular carcinoma, and increased liver-related mortality. Hepatic fibrosis, driven by the activation of hepatic stellate cells (HSCs), is the primary predictor of mortality and adverse liver events in MASH patients. Understanding the molecular mechanisms underlying HSC activation is crucial for developing effective antifibrotic therapies. However, the molecular triggers and drivers of HSC activation remain poorly understood. Roi et al. investigated the role of Transmembrane 7 Superfamily Member 3 (TM7SF3) in fibrosis induced by MASH.
The study successfully established TM7SF3 knockout mouse models and liver organoid models. Antisense oligonucleotide (ASO) intervention experiments were conducted in these mouse models, as well as in human hepatic stellate cells (HSCs) and organoid models. The results showed that in human HSCs, ASO 56 treatment significantly reduced the expression of TEAD1ΔEx5 and decreased the expression of fibrosis-related genes such as Tgfb1, Acta2, Timp1, Il-6, and Pdgfrb. In mouse liver organoid models, ASO 56 treatment inhibited the expression of fibrosis genes and proteins while increasing the expression of the quiescent HSC marker Bambi. In in vivo experiments, a single dose of ASO 56 (10 mg/kg) administered for 7 days significantly reduced the expression of TEAD1ΔEx5 and Cyr61 in mouse HSCs, without significantly affecting hepatocytes and non-parenchymal cells (NPCs). These results indicate that ASO 56 effectively inhibits HSC activation and liver fibrosis by blocking TEAD1 alternative splicing and confirms its efficacy in more complex tissue environments. This validates the potential of antisense oligonucleotide technology in treating fibrotic diseases.
Additionally, quantitative assessment of gene and protein expression demonstrated that TM7SF3 knockout or ASO 56 treatment significantly affected the expression of fibrosis- and inflammation-related genes and proteins. These data include significant changes in the expression of genes such as Tgfb1, Col1a1, Acta2, Timp1, Il-6, and MCP1 in HSCs and liver tissues. These findings provide quantitative evidence for the role of TM7SF3 and TEAD1 alternative splicing in HSC activation and liver fibrosis.
Compared to traditional methods for studying liver fibrosis, the ASO 56 proposed in this study exhibits stronger targeting, enhancing both the precision and efficacy of the intervention. This technology not only shows potential in the MASH model but may also be applicable to other diseases involving alternative splicing. Additionally, the study employed various models to validate the results, thereby increasing the reliability and reproducibility of the conclusions. In summary, this research reveals a new mechanism by which TM7SF3 regulates TEAD1 alternative splicing and introduces a novel antisense oligonucleotide intervention method. It provides new therapeutic insights and strategies for addressing MASH-related liver fibrosis.
Despite the significant progress made in elucidating the mechanism by which TM7SF3 regulates TEAD1 alternative splicing and its role in MASH-related liver fibrosis, several limitations and areas require further validation. For instance, the study primarily focuses on fibrosis regulation, and the comprehensive regulation of inflammation needs further exploration. In particular, more research is needed to understand the role of TM7SF3 in other signaling pathways and its combined effects on inflammation and fibrosis. Additionally, although ASO 56 has demonstrated antifibrotic effects in vivo, its long-term efficacy and safety remain unclear. Moreover, the specificity of ASO 56's effects in HSCs raises questions about potential impacts on other cell types, which is crucial for evaluating its systemic side effects and overall safety.
Overall, future research should explore conditional knockout models, double knockout models, gender differences, ASO absorption and distribution, and cross-species applicability to ensure comprehensive results and broad application prospects. This will aid in developing more effective and safer antifibrotic therapies, addressing global health challenges posed by MASH and other fibrotic diseases.
Isaac, Roi, et al. "TM7SF3 controls TEAD1 splicing to prevent MASH-induced liver fibrosis." Cell Metabolism 36.5 (2024): 1030-1043.





Post comments