
by University of California - Berkeley
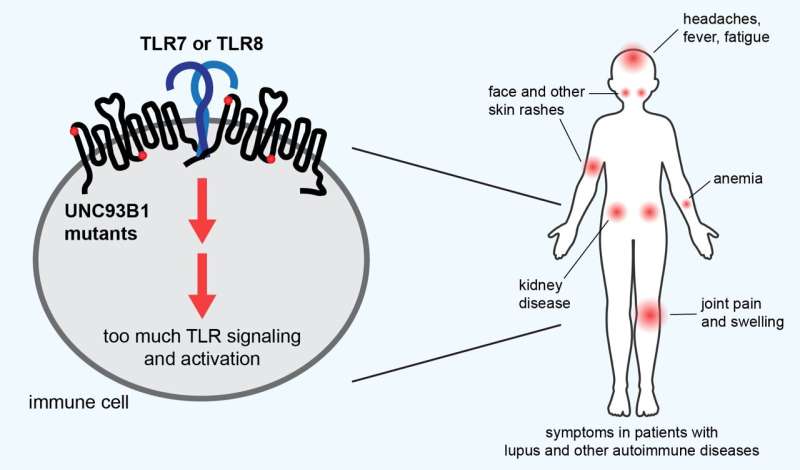
Many autoimmune diseases, including lupus, have been linked to problems with the toll-like receptors (TLR) on immune cells, in particular the TLR7 and TLR8 receptors, which recognize the nucleic acids in RNA from invading viruses and bacteria. While TLRs are critical to mobilizing the body's immune defenses against these invaders, if they are out of tune, they can activate the immune system against the body's own nucleic acids, leading to painful symptoms. Researchers at UC Berkeley have shown that mutations in the UNC93B1 gene, which regulates TLRs, are associated with autoimmune symptoms in mice and humans. Credit: Victoria Rael and Gregory Barton, UC Berkeley
Lupus is a lifelong, often painful and occasionally lethal autoimmune disease. Few treatments exist today beyond powerful steroids to knock down a patient's immune system—a therapy that has its own serious risks.
The good news is that new and promising treatments are in clinical trials. But the term lupus belies the fact that the disease has a variety of causes, which means that treatments will have to be highly personalized to guarantee that each patient is given the drug that targets the specific genetic mutation responsible for their variety of lupus.
Researchers are just now beginning to link specific genetic mutations to subsets of lupus patients, allowing physicians to target therapies to those who will benefit most. In the latest advance, researchers at the University of California, Berkeley, report in a new paper the discovery of two sets of patients with genetic mutations that are nearly identical to mutations that the researchers had earlier pinpointed in mouse and cell lines as linked to autoimmune disease.
These two genetic links are among several dozen mutations that the UC Berkeley team recently discovered and linked to lupus, all in one gene that regulates a prime suspect in a subset of lupus patients—proteins called toll-like receptors (TLR), which enable immune cells to recognize foreign DNA and RNA.
According to study leader Gregory Barton, UC Berkeley professor of molecular and cell biology, identifying these mutations could help doctors deliver a personalized treatment to patients with oversensitive TLRs and, in particular, oversensitive TLR7 receptors.
"We basically have a map now," said Barton, who is also an investigator in the Howard Hughes Medical Institute. "It's not like everybody that has lupus has a mutation in the gene that causes overactivation of TLRs and TLR7. But there are drugs coming online that very specifically inhibit TLR7.
"As we sequence more and more people, it will become easier to identify those patients and put them on those drugs. That's a lot better than the current course of therapy for lupus, which is brutal."
"This is exciting because the drug will be orally available and is in clinical trials now," said Victoria Rael, a UC Berkeley graduate student who, with fellow graduate student Julian Yano, is a co-first author of the paper.
The results of the genetic screens and details of the patients' mutations were published in the Journal of Experimental Medicine.
A problem recognizing 'self'
Autoimmune diseases, which range from rheumatoid arthritis and Crohn's disease to scleroderma and numerous thyroid conditions, stem from attacks by the immune system on the body's own cells that destroy normal, healthy tissue.
Many studies have linked at least two types of autoimmune disease, lupus and psoriasis, to TLRs, which are part of the innate immune system that initially detects foreign invaders, such as viruses and bacteria, and stimulates a first line of attack. Normally, TLRs are delicately tuned to react only to foreign DNA and RNA, but if that tuning is off, they can react to a body's own nucleic acids and proteins associated with nucleic acids, which look much like those of pathogens.
What makes this autoimmune reaction so deadly is that the TLRs also activate the body's second-line defense, the more powerful adaptive immune response, mobilizing T and B cells, macrophages, and other cells. These cells then mount a sustained attack that destroys the body's healthy tissue and causes chronic inflammation.
The most common form, systemic lupus erythematosus (SLE), for example, is characterized initially by skin rashes—in particular, a butterfly-shaped rash on the face—but later by damage to joints, muscles, organs and skin, causing pain and fatigue. It's most commonly seen in females, often starting during the teen years. Lupus, in general, is two to three times more prevalent among women from many ethnic and racial minority groups than among white women.
"We think the way the system works is that if nucleic acids find these receptors, most likely they're going to be from a virus," Barton said. "But in some people, the receptor is more responsive, so now levels of self-nucleic acids that otherwise wouldn't stimulate the receptor in a normal person activate the receptor. We think that one of the ways that these mutations are working is that they're making levels of self-nucleic acids that normally wouldn't be stimulatory, stimulatory."
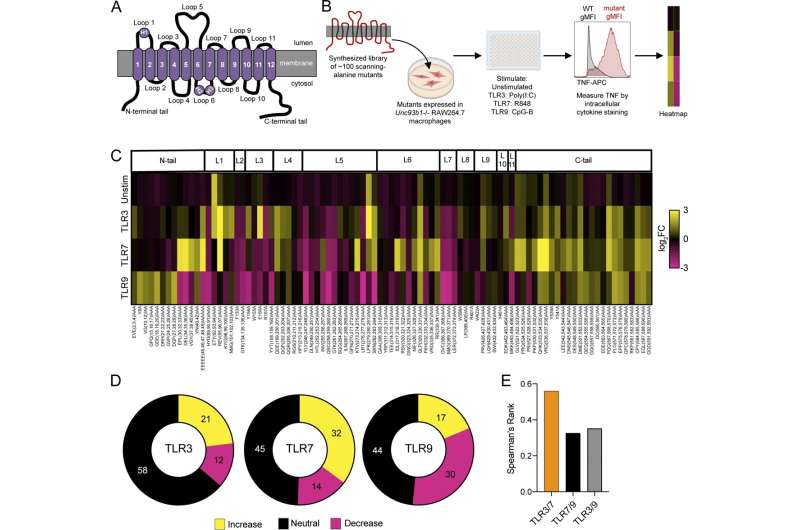
A scanning-alanine mutagenesis screen reveals distinct domains of UNC93B1 that regulate endosomal TLR signaling. Credit: (2024). DOI: 10.1084/jem.20232005
Barton and his lab colleagues have been investigating the role of TLRs that are misregulated in lupus, and in particular, one of the main proteins that regulates them: UNC93B1, or UNC for short. Several years ago, a team of postdoctoral fellows and graduate students in his lab screened in cell culture more than 100 genetic mutations in the UNC gene to see which ones overstimulated TLRs and would be good targets for further study.
While they published some details in earlier papers, they didn't publish the complete list because there seemed to be little point—almost no data was available on the genome sequences of lupus patients to compare with the mutations that overstimulated TLRs.
But that has changed in recent years, thanks to a plunge in the cost of genome sequencing. That's how the mother of a young girl with severe autoimmune disease found Barton. Her daughter's DNA had been sequenced and showed a mutation in a region of UNC that Barton's team had noted in an earlier paper.
Lupus in the family
Rael and undergraduate Madeleine Weiss used the same cell culture screening technique to test the novel mutation from the young girl and found that it had an overstimulating effect, similar to the effect of other mutations in that area of the UNC gene. Surprisingly, the patient had the genetic mutation on only one of the two UNC alleles, meaning that she had one normal UNC gene, yet she still suffered severe autoimmune symptoms.
Barton and his team also connected with a family of five afflicted with lupus. All had mutations on one UNC allele in another area of the UNC protein that Barton's team had previously identified. That mutation, when screened in cell lines, also produced overactive TLRs.
"We were skeptical that just one copy of a gene would be sufficient to cause a disease," Rael said. "It wasn't until we put the patients' mutations into cell lines and saw that they led to very convincing TLR hyper-responsiveness that we realized they had the possibility of being sufficient to be disease-causing."
Rael and Yano then repeated the screening work previously performed in the lab and confirmed that 32 distinct mutations in the UNC gene—about one-third of the mutations tested—increased the sensitivity of TLR7 to nucleic acids at least twofold. About another five mutants increased TLR7 sensitivity but to a lesser degree.
Before these screens, only two mutations in the UNC protein had been linked to increased sensitivity of TLR7 in mice, though three additional human mutations were reported within the last two months.
Barton is hopeful that by publishing the complete list of TLR hypersensitivity mutations, doctors can identify other lupus patients who could benefit from the anti-TLR drugs now in clinical trials. One drug, M5049, or Enpatoran, appears to work by latching onto two human receptors, TLR7 and TLR8, and preventing them from binding nucleic acids.
Rael, Yano and other members of Barton's lab are investigating further how these unique UNC mutations affect the way a patient manifests the disease. They have recreated these patients' mutations in mice so that they can model human lupus.
"With mouse models, you can start thinking about how, even though the mutations are in the same protein, the different mechanisms of TLR regulation break down, which immune cells get activated as a result, and how this can lead to differences in the symptoms patients suffer from," Rael said.
The lab also is trying to understand how UNC tunes TLRs, which may be by regulating the number and arrangement of TLRs on immune cells. More TLRs may make a person more sensitive to the small number of self-nucleic acids circulating in the body.
"UNC93B1 is important for getting the receptors to the place where they can function, but it also is important for regulating them when they get there," Barton said. "The protein is a very baroque way of trying to make decisions about whether the nucleic acid that you just bound to a TLR is from a virus or from one of your own cells."
He hopes that physicians add this gene to the list of lupus-associated genes, "so if they see a mutation like these, even a heterozygous mutation, they will investigate further."
Other senior authors of the paper are Bo Liu of the Chinese Academy of Sciences in Shanghai and Olivia Majer of the Max Planck Institute for Infection Biology in Berlin, Germany. The co-authors also include physicians from UC San Francisco, Stanford University, and hospitals in Missouri, North Carolina and Washington.
More information: Victoria Rael et al, Large scale mutational analysis identifies UNC93B1 variants that drive TLR-mediated autoimmunity in mice and humans, Journal of Experimental Medicine (2024). DOI: 10.1084/jem.20232005
Journal information: Journal of Experimental Medicine
Provided by University of California - Berkeley





Post comments