
Today, we are introducing a paper from the team led by E. A. Huerta. Predicting the three-dimensional structure of proteins from their amino acid sequences is a major computational challenge in biophysics and plays a critical role in robust protein structure prediction algorithms, with applications ranging from drug discovery to genome interpretation. With the emergence of AI models like AlphaFold, applications relying on robust protein structure prediction algorithms are undergoing a revolutionary transformation. To maximize the impact of these AI tools and simplify their use, the authors introduce APACE (AlphaFold2 coupled with supercomputing), a computational framework that efficiently handles the AI model and its terabyte-scale databases, enabling accelerated protein structure prediction analysis in modern supercomputing environments. The authors deployed APACE on the Delta and Polaris supercomputers and quantified its performance in accurate protein structure prediction using four typical proteins (6AWO, 6OAN, 7MEZ, and 6D6U). Utilizing up to 300 combinations distributed across 200 NVIDIA A100 GPUs, the authors found that APACE was up to two orders of magnitude faster than the out-of-the-box AlphaFold2 implementation, reducing the solution time from weeks to minutes. This computational approach can easily be integrated with robotic laboratories to automate and accelerate scientific discovery.

Innovations combining artificial intelligence and supercomputing are driving breakthroughs in science and engineering. The rise of AI models like GPT-4 and AlphaFold provides new capabilities for accelerating and automating scientific discovery. However, some of these models are not publicly available, breaking an important tradition within the AI community. It is said that the vast scale of these AI models makes them difficult for many potential users to access.
To address this issue, the authors demonstrate how large AI models can be integrated with high-performance computing platforms to help a broad user base fully harness AI's capabilities for scientific discovery. The authors chose AlphaFold2 as the scientific driver for this study because this AI model is revolutionizing discoveries in the field of biophysics, and its application in accurate and rapid protein structure prediction (PSP) requires optimal utilization of modern supercomputing environments. Here, the authors show how to optimize AlphaFold2 and its over 2.6 TB database to reduce the time needed for accurate PSP from weeks to minutes.
Key Features of AlphaFold2: AlphaFold2 uses central processing units (CPUs) to compute key input features: multiple sequence alignments (MSA) and structural templates. MSAs represent collections of protein sequences homologous to the query protein. MSAs capture evolutionary relationships between different proteins, such as conserved and variable amino acid residues. MSAs are calculated using CPU-based sequence alignment algorithms like Jackhmmer, which aligns the query protein sequence with known homologous sequences retrieved from databases like Uniclust. AlphaFold2 can extract critical residue interactions from the MSA. Additionally, structural templates refer to experimentally known homologous protein structures that share significant sequence similarity with the query protein. These templates are used to enhance AlphaFold2's prediction accuracy. CPU-based algorithms like HHsearch (for monomers) and Hmmsearch (for multimers) are used to search public protein structure databases, such as the Protein Data Bank (PDB). AlphaFold2 then extracts spatial information from the templates relevant to the query protein.
In the GPU stage, AlphaFold2 processes the features generated from MSAs and templates through the Evoformer network. The Evoformer iteratively exchanges information between the MSA and pairwise interactions to extract relationships between amino acid residues. The updated representation enters the structure module, where predictions for the rotation and translation of each residue's position are made.
The generated predicted 3D structure is relaxed through a minimization process using a molecular dynamics (MD) engine to enhance accuracy. Once the final structure is generated, the information loops back to the starting point of the Evoformer module to further optimize the structural prediction. Overall, AlphaFold2 is trained end-to-end with exceptional accuracy and reliability, capable of predicting the three-dimensional structure of proteins.
Improvements to AlphaFold2 with APACE: The authors introduced APACE, a service combining AlphaFold2 and supercomputing, which is a computational framework that accelerates AlphaFold2 through CPU and GPU optimization and distributed computing in supercomputing environments. Key features of this approach include:
Data Management: First, APACE simplifies the use of AlphaFold2 by hosting the 2.6 TB AI model and database on the Delta and Polaris supercomputers. AlphaFold2's neural networks can easily access data by utilizing solid-state drive (SSD) data storage and Infinite Memory Engine (IME) data tiering.
CPU Optimization: APACE uses the Ray library for CPU optimization to parallelize CPU-intensive MSA and template computations. As part of CPU optimization, APACE allocates more CPU cores to MSA/template search tools than the default 4 or 8 cores, which showed heuristic speed improvements in experiments. Additionally, the authors implemented a checkpoint to skip redundant MSA/template steps if a features.pkl file (an intermediate file storing MSA/template search results) already exists.
GPU Optimization: Third, APACE uses the Ray library for GPU optimization to parallelize the GPU-intensive neural network protein structure prediction steps. Unlike ParaFold’s GPU acceleration, which uses only model_1 (a template-based pre-trained model) to predict one conformation for peptide sequences (typically less than 100 amino acid residues), APACE predicts multiple conformations for each protein sequence, using all five pre-trained models in parallel when necessary, which is more computationally demanding.
New Features: Fourth, APACE can predict multiple monomer conformations for each pre-trained neural network model (a total of five models), a feature that was only available in multimer prediction in the original AlphaFold2 model. APACE also includes additional features such as enabling dropout during structure prediction, altering the number of Evoformer iterations, or sub-sampling MSA options.
The authors completed three computational experiments to compare the performance of APACE with the original AlphaFold2 model in detail. Each experiment will be described in turn, along with the corresponding results.
Experiment 1: Predicting the Structure of Four Benchmark Proteins
The authors selected four proteins as benchmarks to evaluate the effectiveness and operational performance of APACE. To predict protein structures using APACE, the authors developed scientific software that allows users to provide appropriate headers in sbatch scripts and load the correct environment and modules, enabling successful simulation submissions on the Delta and Polaris supercomputers.
Monomer Proteins: The authors used the monomer protein 6AWO (serotonin transporter) as a basic structure and tested the accuracy and conformational diversity of the baseline prediction using five models. To do this, the authors created a Ray cluster consisting of eight NVIDIA A100/A40 GPUs (equivalent to two A100/A40 GPU nodes in Delta and Polaris) to facilitate parallel CPU and GPU execution and relaxation optimization for all five models (one structure per model).
Multimer Proteins: For multimer proteins, the authors tested 6OAN (Duffy binding protein bound to a single-chain variable fragment antibody), 7MEZ (phosphatidylinositol 3-kinase), and 6D6U (a pentameric GABA transporter protein with three different chains), which represent more challenging cases in multimer prediction. For each protein, the authors conducted eight structure predictions per model, generating a total of 40 predictions (five model sets × eight predictions per model). To simultaneously execute and relax all 40 models, a Ray cluster was used with 40 NVIDIA A100 and 40 A40 GPUs (10 A100/A40 GPU nodes).
CPU Acceleration: By implementing parallel optimization techniques, APACE achieved an average CPU acceleration of 1.8x on the Delta supercomputer and 1.78x on the Polaris supercomputer. These results were independent of the number of computational nodes.
GPU Acceleration: APACE also showed significant improvements in GPU acceleration. The following results were obtained using 8 GPUs for 6AWO and 40 GPUs for 6OAN, 7MEZ, and 6D6U:
6AWO: On the Delta supercomputer, A40 and A100 GPUs achieved 4.4x and 4.98x acceleration, respectively, on Polaris.
6OAN: On the Delta supercomputer, A40 and A100 GPUs achieved 34x and 32.4x acceleration, respectively, and 40.1x acceleration on Polaris.
7MEZ: On the Delta supercomputer, A40 and A100 GPUs achieved 37.5x and 37.1x acceleration, respectively, and 40x acceleration on Polaris.
6D6U: On the Delta supercomputer, A40 and A100 GPUs achieved 21.2x and 22x acceleration, respectively, and 40x acceleration on Polaris.
Table 1: Performance Benchmark Between Out-of-the-Box AlphaFold2 and the APACE CPU & GPU Optimized Framework

The authors summarized these results in Table 1. It is noteworthy that prediction times were consistently shorter when using NVIDIA A100 GPUs. In short, APACE provides significant acceleration for both basic and complex structures while maintaining the accuracy and robustness of the original AlphaFold2 model.
Experiment 2: Predicting Protein 7MEZ Using 100 and 200 NVIDIA A100 GPUs
To quantify the performance and scalability of APACE on the Delta and Polaris supercomputers, the authors conducted predictions for protein 7MEZ using a large number of computational nodes. Specifically, the authors utilized 100 NVIDIA A100 GPUs, equivalent to 25 A100 GPU compute nodes, to generate predictions (20 predictions per model). Similarly, the authors leveraged the computational power of 200 NVIDIA A100 GPUs, equivalent to 50 A100 compute nodes, generating a total of 200 predictions (40 predictions per model).
Table 2: Performance Benchmark Between Out-of-the-Box AlphaFold2 and APACE for Protein 7MEZ

As shown in Table 2, APACE achieved significant acceleration. When the authors computed 100 sets of 7MEZ using 100 GPUs, APACE completed the required calculations in 67.8 minutes, whereas AlphaFold2 on the Delta supercomputer required 6068.8 minutes (101.1 hours/4.2 days). On the Polaris supercomputer, APACE reduced the solution time from 8793.3 minutes (146.5 hours/6.1 days) to 87.9 minutes.
Similarly, if the authors needed to compute 200 sets for the same protein, APACE completed all predictions in 64 minutes on the Delta supercomputer, whereas the original AlphaFold2 method required 12023.3 minutes (200.4 hours/8.3 days). On the Polaris supercomputer, APACE reduced the solution time from 12741.2 minutes (212.4 hours/8.8 days) to just 84.9 minutes.
Finally, when predicting 300 sets of the 7MEZ protein, APACE completed all predictions in 68.2 minutes on the Delta supercomputer, whereas the original AlphaFold2 method required 18064.3 minutes (301.1 hours/12.5 days). On the Polaris supercomputer, APACE reduced the solution time from 15295.6 minutes (254.9 hours/10.6 days) to just 76.9 minutes.
Experiment 3: Diversity of APACE Ensembles
The inherent limitations of AlphaFold2 only allowed the authors to generate five predictions per monomer, with one prediction per model, limiting the diversity of protein conformations. Additionally, fine-tuning parameters such as dropout were not feasible. However, the authors successfully overcame this limitation by modifying ColabFold's code.
APACE allows users to choose the following options:
Set the number of ensembles in the structure module using -num_ensemble.
Control the number of recycles with --num_recycles.
Subsample MSAs using --max_seq and --max_extra_seq.
Perform Evoformer fusion with --use_fuse.
Enable Bfloat16 mixed precision calculations with --use_bfloat16.
Use --use_dropout to perform diversified conformation sampling based on a Bernoulli mask.
APACE's Protein Structure Prediction and Conformation Diversity: The authors modified AlphaFold2’s code to reflect the flexible protein structure prediction parameter customization seen in ColabFold. Through these improvements, the authors successfully extended the range of predictions, thereby enhancing the overall reliability of the predicted structures. While protein structure prediction is of great importance, the authors aim to extend APACE to predict conformation diversity, as proteins are not static but rather dynamic and flexible structures. Sampling from a broad range of conformations is crucial for drug discovery.

Figure 1
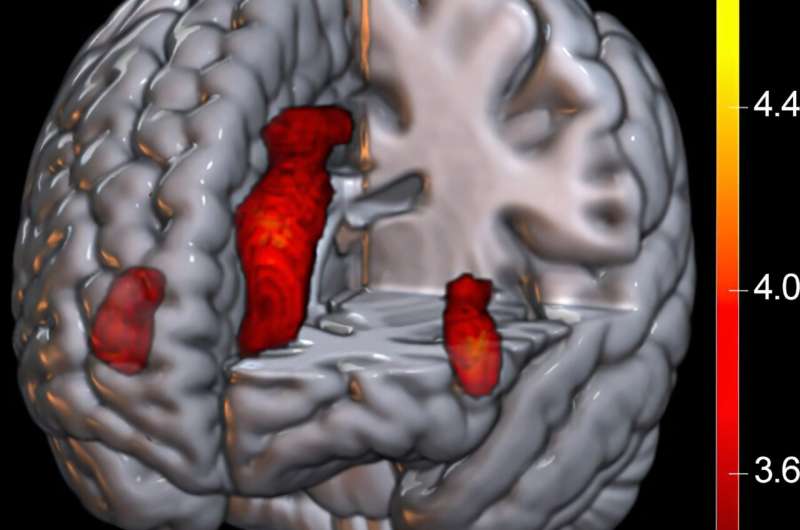
In the case of 6AWO (approximately 500 amino acid residues), as shown in Figure 1, the authors enhanced the customization of parameters (choosing the option --use_dropout=True) and predicted 100 structures of the serotonin transporter (SERT). The authors found that the structures predicted by APACE were comparable to the real structure. When visualizing the transmembrane α-helices with high variation (the cyan-colored parts in the right figure), TM2, TM6, TM10, and TM12 were prominently highlighted. Among these, TM6, TM10, and TM12 are responsible for conformational changes or ligand binding from the outside to the inside, indicating that APACE has learned to predict a wide range of conformations for SERT. APACE’s predictions for SERT were accurate, with SERT being a complete membrane protein. Even in the absence of the membrane, APACE was able to accurately predict the transmembrane domains, demonstrating its potential in drug discovery research.

Figure 2

Figure 3
For 6OAN, 7MEZ, and 6D6U (approximately 600, 2000, and 1800 amino acid residues, respectively), the authors conducted multimer predictions. Figures 2 and 3 show that both 6OAN and 7MEZ accurately predicted the conformational ensembles of heterodimer structures. Specifically, the predicted interface binding poses were comparable to the real structures, although there may be minor errors in the predicted secondary structure that are unrelated to the interface binding. However, the correctly predicted interface binding poses between proteins, through non-bonded interactions, are more important. These minor errors can be addressed through molecular dynamics, Monte Carlo methods, and protein design tools.

Figure 4
In Figure 4, for 6D6U, the predicted structures comparable to the real structure (left image) can be observed, as well as incorrect structures caused by errors in predicting the homodimer positions (right image). Since 6D6U is a membrane pentameric heteroprotein, predicting the correct structure of each monomer and the alternating chain pattern is a challenging task. Thus, the prediction of transmembrane helices might be inaccurate, but the overall structure still aligns fairly well with the real structure.
In summary, the authors demonstrated APACE’s capabilities in predicting protein structures, reflecting the robustness and accuracy of AlphaFold2, while providing significant acceleration—reducing solution time from days to minutes. However, APACE may face limitations when predicting transmembrane proteins and/or multichain multimers, which are inherited from AlphaFold2.
Conclusion
The authors introduced APACE, a framework that retains the robustness and accuracy of AlphaFold2 while leveraging supercomputing technology to reduce the time from data to insights from several days to just a few minutes. The authors achieved this by: a) efficiently utilizing the data storage and tiering systems of the Delta and Polaris supercomputers; b) optimizing CPU and GPU computational performance; and c) developing scientific software that enables the prediction of protein structure conformational ensembles. These tools are released alongside this paper, providing researchers with a computational framework that can be easily integrated with robotic laboratories to automate and accelerate scientific discovery.
References
Park H, Patel P, Haas R, et al. APACE: AlphaFold2 and advanced computing as a service for accelerated discovery in biophysics[J]. Proceedings of the National Academy of Sciences, 2024, 121(27): e2311888121.



Post comments