
Credit:Dynamic metabolism of endothelial triglycerides protects against atherosclerosis in mice.
Atherosclerosis, the buildup of plaque in arteries, remains a leading cause of cardiovascular disease and death. While traditional risk factors such as cholesterol and blood pressure are well-established, recent research has highlighted the critical role of metabolism in plaque formation and progression. Two recent studies provide valuable insights into how alterations in metabolic pathways contribute to plaque instability and vulnerability.
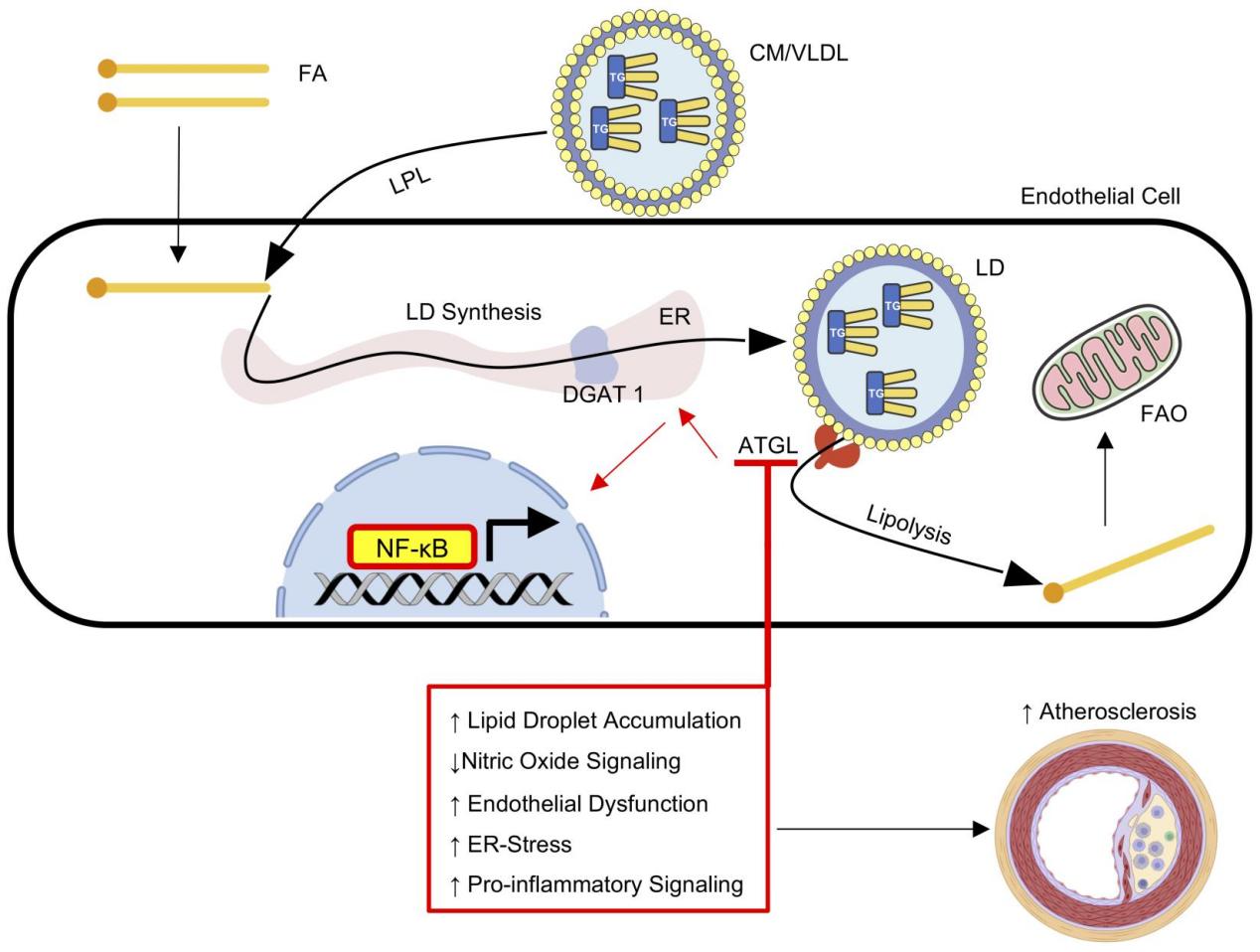
The first study delved into the intricate relationship between endothelial triglyceride metabolism and large vessel function, with a specific focus on the pivotal role of adipose triglyceride lipase (ATGL). Employing a multifaceted approach encompassing genetically modified mice, cell culture experiments, and RNA sequencing, the authors unveiled a complex interplay of lipid metabolism, inflammation, and endothelial dysfunction that ultimately contributed to the development of atherosclerosis.
The study began with the creation of endothelial-specific ATGL knockout mice (Atgl ECKO), a groundbreaking model that allowed for a precise investigation of ATGL's function within the endothelium. These mice exhibited a striking accumulation of neutral lipids and lipid droplets (LDs) in both micro- and macrovessels, reminiscent of the lipid-rich environment found in human atherosclerotic plaques. The observation of LD accumulation in the absence of systemic lipid abnormalities suggested that endothelial triglyceride metabolism played a crucial role in maintaining vascular homeostasis and preventing lipid-induced damage.
To further explore the functional consequences of ATGL deficiency, the authors assessed the vasomotor function of Atgl ECKO mice. The results were alarming, revealing impaired endothelium-dependent vasorelaxation and reduced nitric oxide (NO) synthesis in aortic segments. This dysfunction was attributed to decreased endothelial nitric oxide synthase (eNOS) protein levels and reduced bioavailability of NO, highlighting the critical role of ATGL in maintaining endothelial function and vascular tone.
The link between ATGL deficiency and atherosclerosis was further solidified through the development of an atherosclerosis model using Atgl ECKO mice crossed with ApoE-deficient mice and fed an atherogenic diet. These mice exhibited a significant acceleration of atherosclerosis development, with larger lesions in the aorta and other vessel segments. The atherosclerosis-prone regions of the aorta showed increased macrophage infiltration, further corroborating the proinflammatory phenotype observed in the endothelium.
To gain mechanistic insights into the observed endothelial dysfunction and inflammation, the authors performed RNA sequencing of LECs from Atgl ECKO mice. The analysis revealed significant alterations in gene expression, including upregulation of inflammatory signaling pathways and ER stress-related genes. The heightened expression of proinflammatory genes, such as VCAM1 and COX2, and the increased responsiveness to inflammatory stimuli like TNF-α and palmitate further support the proinflammatory phenotype of Atgl ECKO endothelial cells.
The authors proposed that ER stress, triggered by ATGL deficiency, played a key role in mediating the proinflammatory response and endothelial dysfunction observed in Atgl ECKO mice. This hypothesis was supported by the findings that ER stress markers like ATF4 and CHOP were upregulated in Atgl ECKO LECs, and that inhibition of ER stress using a chemical chaperone (4-PBA) partially rescued the proinflammatory phenotype and the heightened responsiveness to palmitate.
The precise mechanism by which ATGL deletion induced ER stress remained an intriguing question. The authors proposed several possibilities, including impaired triglyceride hydrolysis leading to altered mitochondrial function and calcium signaling, ultimately triggering ER stress in endothelial cells. Further investigation into the metabolic pathways downstream of ATGL deficiency is necessary to fully understand the molecular mechanisms linking triglyceride metabolism and ER stress in the endothelium.
Credit: Genome-scale metabolic network of human carotid plaque reveals the pivotal role of glutamine/glutamate metabolism in macrophage modulating plaque inflammation and vulnerability
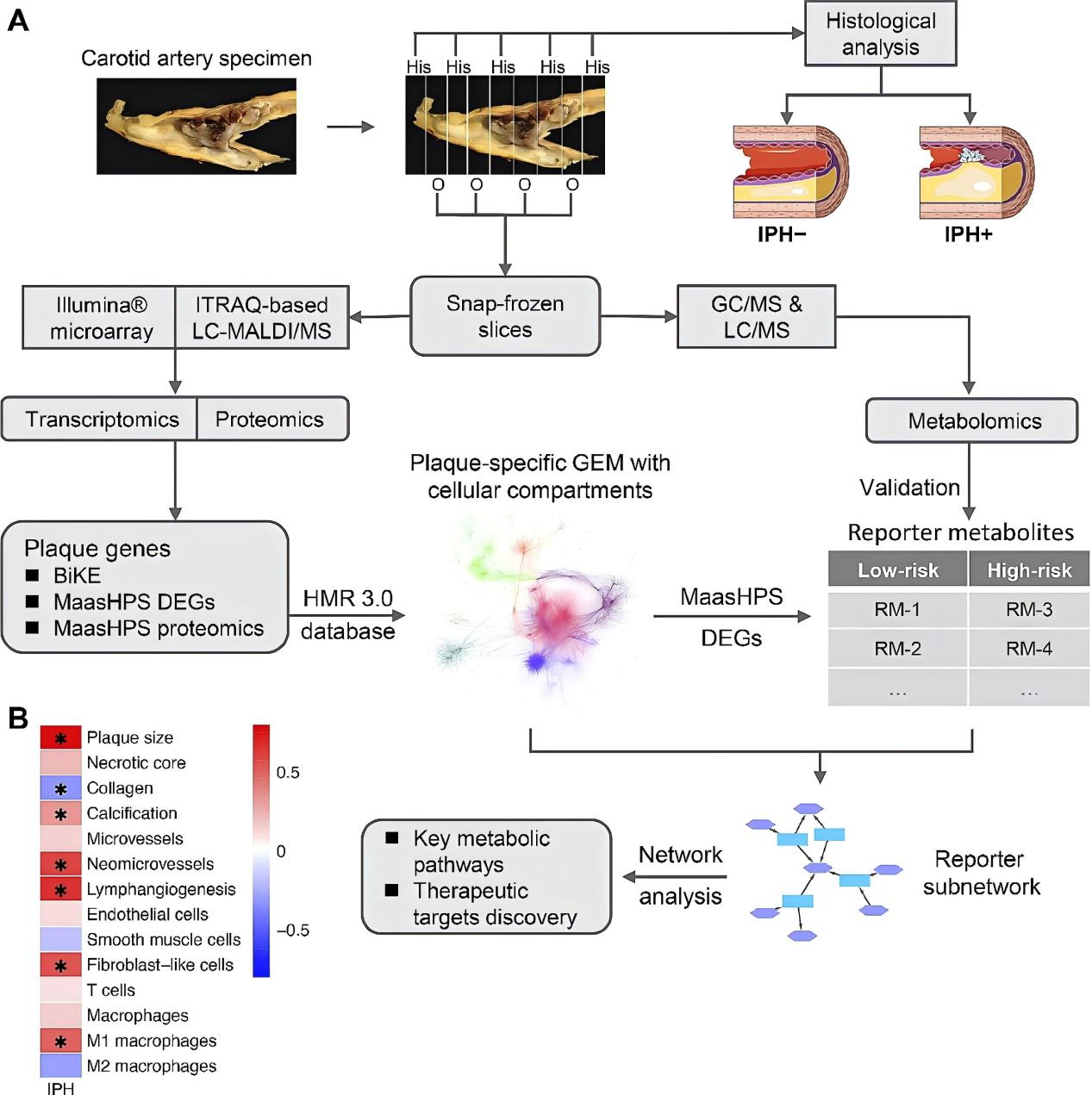
Schematic workflow. A The entire carotid endarterectomy specimen was cut in parallel 5 mm thick slices, snap-frozen in liquid nitrogen and stored until use. Every second slice was sectioned. After H&E staining, sections were categorized and classified histologically based on the presence/absence of IPH, a proxy of plaque stability. Sections were pulverized and aliquoted for transcriptomics, proteomics, and metabolomics analyses. Highly expressed genes in the BiKE cohort were aggregated with the MaasHPS DEGs and proteomics for building the plaque-specific GEM, after which the MaasHPS DEGs were used to infer the reporter metabolites from the well-annotated plaque-specific GEM, which were subsequently validated by the MaasHPS metabolomics. The reporter subnetwork evolved from the GEM and was used to analyze key pathways and potential therapeutic targets for plaque stability. B Spearman’s correlation between IPH area and other plaque traits. *P-value < 0.05.
In addition to triglyceride metabolism in endothelial cells, the second study conducted by Jin et. al. focused on glutamine/glutamate (Gln/Glu) metabolic pathway in macrophages, which was associated with the transition of atherosclerotic plaques from stable to unstable hemorrhagic states. By integrating transcriptomics, proteomics, and metabolomics data from human carotid artery plaques, the researchers constructed a comprehensive genome-scale metabolic network (GEM) specific to atherosclerotic plaques. This GEM revealed significant alterations in lipid, cholesterol, and inositol metabolism, along with increased lysosomal activity and inflammatory response in unstable plaques with intraplaque hemorrhage (IPH+).
One of the key findings was the dysregulation of the Gln/Glu metabolic pathway in IPH+ plaques. The conversion of glutamine to glutamate and their flux between the cytoplasm and mitochondria were compromised, leading to a significant reduction in overall glutamate levels. This dysregulation was strongly associated with macrophage content and a pro-inflammatory phenotype in IPH+ plaques, suggesting an inflammation-prone microenvironment.
To further validate these findings, the researchers performed single-cell RNA sequencing analysis of human plaques. This analysis confirmed that the Gln/Glu pathway genes were predominantly expressed in macrophages, particularly in the IPH+ plaques. Furthermore, the expression of key genes involved in Gln/Glu metabolism, such as GLUL and SLC25A12, was significantly correlated with plaque macrophage content.
To investigate the functional implications of Gln/Glu pathway dysregulation, the researchers conducted experiments on human macrophages. They found that glutamine deprivation or GLUL inhibition affected various macrophage functions, including apoptosis, mitochondrial stress, lipid uptake, phagocytosis, inflammasome activation, and cell morphology. These findings suggested that the altered Gln/Glu pathway and glutamate availability have a profound impact on macrophage functions and phenotype, contributing to plaque instability.
Overall, these studies provide compelling evidence that alterations in metabolic pathways contribute significantly to plaque instability and vulnerability. Understanding the complex interplay between metabolism and atherosclerosis is crucial for developing novel therapeutic strategies to prevent plaque rupture and reduce cardiovascular events.
References:
Boutagy NE, Gamez-Mendez A, Fowler JW, Zhang H, Chaube BK, Esplugues E, Kuo A, Lee S, Horikami D, Zhang J, Citrin KM, Singh AK, Coon BG, Lee MY, Suarez Y, Fernandez-Hernando C, Sessa WC. Dynamic metabolism of endothelial triglycerides protects against atherosclerosis in mice. J Clin Invest. 2024 Jan 4;134(4):e170453.
Jin H, Zhang C, Nagenborg J, Juhasz P, Ruder AV, Sikkink CJJM, Mees BME, Waring O, Sluimer JC, Neumann D, Goossens P, Donners MMPC, Mardinoglu A, Biessen EAL. Genome-scale metabolic network of human carotid plaque reveals the pivotal role of glutamine/glutamate metabolism in macrophage modulating plaque inflammation and vulnerability. Cardiovasc Diabetol. 2024 Jul 8;23(1):240.






Post comments