
by Kyoto University
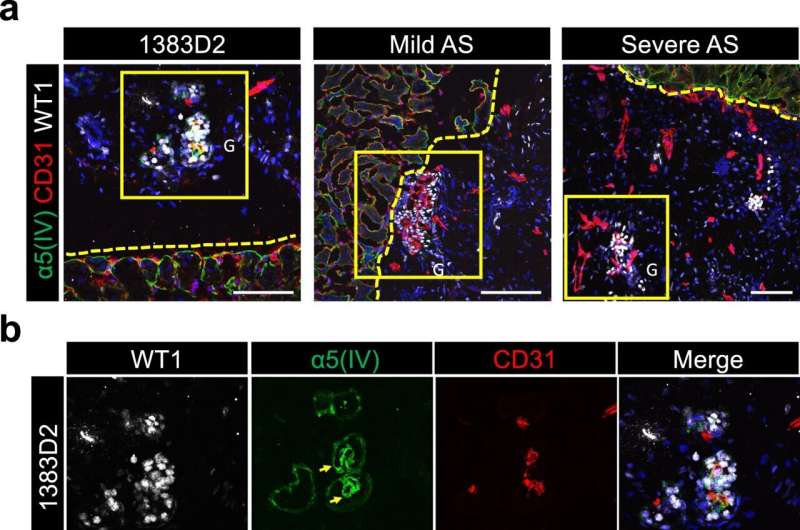
Patient iPSC-derived kidney organoids become vascularized and recapitulate AS phenotypes after transplantation in vivo. a Immunostaining images of glomerulus-like structures in day 21 grafts for WT1 (podocytes), α5(IV), and CD31 (mouse endothelial cells). Mouse kidney and grafts from NPCs are divided by the yellow dotted lines. Scale bars, 100 µm. b Magnified images of the yellow boxed areas in a. Arrows indicate α5(IV)-expressing GBM-like structures. G glomerulus. Scale bars, 50 µm. Credit: Communications Biology (2023). DOI: 10.1038/s42003-023-05203-4
Researchers have generated induced pluripotent stem cell (iPSC)-derived kidney organoids to model Alport syndrome caused by a mutation in the COL4A5 gene and have demonstrated the utility of a chemical chaperone to rescue the protein misfolding and collagen misassembling caused by the mutation. The research is published in the journal Communications Biology.
Alport syndrome (AS) is estimated to affect 1 in 5,000 to 1 in 50,000 in the United States and Europe, respectively, and about 1,200 people in Japan are estimated to have the disorder. It is the second most common form of hereditary glomerulonephritis, caused by mutations in several genes (COL4A3, COL4A4, or COL4A5) that form a particular type of collagen (i.e., α3α4α5[IV] collagen) in the glomerular basement membrane (GBM) of the kidney and manifest into end-stage renal disease (ESRD) early in life.
Unfortunately, there is no curative treatment, and the pathogenic mechanism is not fully understood due to a lack of in vitro models to deduce and correct the defects in collagen assembly in AS patients.
A team of researchers from CiRA and Taisho Pharmaceutical Co., Ltd. headed by Professor Kenji Osafune (Department of Cell Growth and Differentiation) created iPSCs from two X-linked AS patients to produce kidney organoids. Specifically, a team of doctors from Kobe University collected blood samples from a patient with mild AS caused by a G545D missense mutation in the COL4A5 gene and another with a severe case of AS caused by a T552Sfs*6 frameshift mutation that produces a truncated collagen α5(IV) protein for iPSC generation for the study.
The research team also produced an AS mutation-corrected iPSC line using the CRISPR/Cas9 genome editing system to reverse the COL4A5 G545D missense mutation in the mild AS patient. Based on a previously reported differentiation protocol with some modifications, they generated kidney organoids through patient-specific and healthy control iPSC-derived nephron progenitor cells.
Notably, the researchers observed that free-floating cultures of the kidney organoids with mechanical agitation helped promote their maturation, a critical feature for this study because GBM collagen composition switches from α1α1α2(IV) to α3α4α5(IV) heterotrimers during kidney development.
As a result, the researchers were able to produce kidney organoids expressing α5(IV) collagen and observed collagen α3α4α5(IV) in the GBM-like structures of both healthy and mutation-corrected mild AS iPSC-derived kidney organoids. Mirroring clinical phenotypes, however, mild and severe AS iPSC-derived kidney organoids expressed α2(IV) but not α5(IV) collagen.
Further analysis by transmission electron micrography revealed that both mild and severe AS kidney organoids failed to show some characteristic GBM ultrastructural abnormalities previously observed in adult AS model (Col4a5 G5x mutant) mice, suggesting further maturation may be necessary for such ultrastructural changes to manifest fully.
Nonetheless, because the kidney organoids had reached a developmental stage to recapitulate the isoform switching of type IV collagen, the researchers could distinguish differences between kidney organoids generated from patient-specific iPSCs originating from mild and severe AS cases. Specifically, COL4A5 expression was only reduced in severe AS kidney organoids, similar to downregulation observed in Col4a5 mutant mice, possibly contributed by nonsense-mediated decay (NMD) of the disease-associated transcript.
However, the mild AS-causing G545D mutation is unlikely to trigger NMD, and thus, COL4A5 expression was unaffected in kidney organoids generated from the mild AS iPSCs. Furthermore, whereas α3(IV) collagen was detected in mild AS kidney organoids, it was absent in those modeling severe AS despite normal expression at the RNA level. Previous clinical studies have shown detectable α3(IV) or α5(IV) protein in AS patients displaying mild symptoms, thus illustrating that the kidney organoids can accurately model various aspects of mild and severe AS pathology.
Collagen heterotrimers normally assemble in the endoplasmic reticulum (ER) and are secreted by the Golgi apparatus into the extracellular space. Defects in assembly, caused by protein misfolding, cause ER stress and trigger autophagic degradation of the unassembled or misassembled collagens.
Because a previous study had demonstrated the potential of chemical chaperones to reduce intracellular collagen accumulation and ER stress in patient primary dermal fibroblasts caused by a COL4A2 mutation by alleviating protein misfolding, the researchers investigated whether a similar approach could work to reverse some of the abnormalities observed in the kidney organoids caused by AS COL4A5 mutations.
To test this, they treated mild AS kidney organoids with several low molecular weight chemical chaperones, 4-phenyl butyric acid (4-PBA), trimethylamine N-oxide (TMAO), and mannitol, and observed α5(IV) collagen in mutant GBM-like structures treated with 4-PBA, thus highlighting its potential as a viable therapeutic strategy for AS treatment.
In summary, this collaborative effort successfully generated an in vitro disease model to reproduce several crucial aspects of AS pathology. Equipped with this new experimental system, researchers can now elucidate the detailed pathogenic mechanisms underlying this debilitating disease and predict the treatment efficacy in response to novel therapies.
More information: Ryuichiro Hirayama et al, iPSC-derived type IV collagen α5-expressing kidney organoids model Alport syndrome, Communications Biology (2023). DOI: 10.1038/s42003-023-05203-4
Journal information: Communications Biology
Provided by Kyoto University





Post comments