
by Tufts University
Credit: EMBO Molecular Medicine (2024). DOI: 10.1038/s44321-024-00110-5
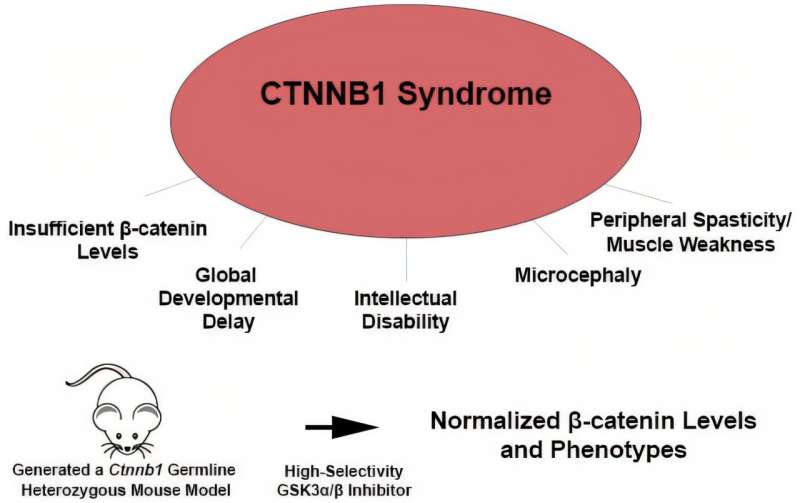
Researchers at Tufts University School of Medicine and the Graduate School of Biomedical Sciences (GSBS) have identified a small molecule that, in mouse and human cell models, rectifies the underlying molecular cause of a rare genetic developmental disorder linked with motor and intellectual disabilities and some types of autism spectrum disorder.
Their most recent research, published in EMBO Molecular Medicine, provides the first evidence that an efficacious drug treatment can be developed for this disorder, called CTNNB1 syndrome, potentially mitigating and reversing the cognitive and motor problems it causes, which can require lifelong assistance and care.
First identified in 2012, CTNNB1 syndrome affects an estimated 1 in 50,000 people worldwide. It is not inherited from parents, but instead is caused in utero or after birth by a sporadic mutation in a single gene. Children at birth look normal, but over time they don't hit expected cognitive and physical milestones. Their heads stop growing, and language delays, learning disabilities, and problems with vision, balance, sitting up, crawling, and walking occur. Some also have autism.
For many years, Michele H. Jacob, professor of neuroscience at the School of Medicine and GSBS, and colleagues have been studying the CTNNB1 gene, which encodes for production of beta-catenin protein, which is critical for proper cell development and function.
"Aberrant levels of beta-catenin have been linked to a high risk of intellectual disability, autism spectrum disorder, and other conditions," says Jacob.
In the most recent research, the scientists developed a mouse model of CTNNB1 syndrome that displays key features of CTNNB1 syndrome in patients and identified a corrective treatment. They show that a small molecule inhibitor of an endogenous enzyme that negatively regulates beta-catenin is effective at bringing the levels up to normal, but not too high. Young adult mice treated with the inhibitor exhibited significantly improved muscle strength and their cognitive abilities returned to levels of healthy mice.
"Seeing these results in young adult mice that are symptomatic suggests that this small molecule could offer important hope to reverse the disease in children with the condition," says Jacob, who has been working regularly with the non-profit patient advocacy group for this disorder, CTNNB1 Connect and Cure, to learn more about the disease.
"We have been testing the small molecule in human cell models of the disease, and recently in patient-derived cells, and it works to increase beta-catenin levels," Jacob says. Tufts and the Broad Institute have jointly filed a patent related to this research, as the effective small molecule was developed at the Broad.
The Tufts scientists are now collaborating with the National Institutes of Health, scientists at the Broad Institute, and colleagues at Boston Children's Hospital to transform the small molecule into a safe and effective drug and develop a pilot clinical trial with patients.
Jacob notes that while 500 children have been definitively confirmed through genetic testing as having CTNNB1 syndrome, the condition may be more widespread than originally thought. Scientists and clinicians are now looking back at cases characterized as cerebral palsy attributed to a lack of oxygen to the brain and are finding that a subset is actually CTNNB1 syndrome.
More information: Jonathan M Alexander et al, Inhibition of GSK3α,β rescues cognitive phenotypes in a preclinical mouse model of CTNNB1 syndrome, EMBO Molecular Medicine (2024). DOI: 10.1038/s44321-024-00110-5
Journal information: EMBO Molecular Medicine
Provided by Tufts University




Post comments