
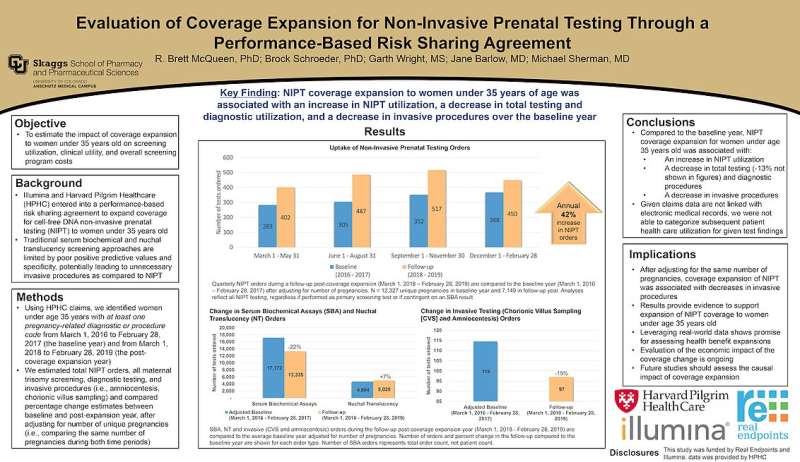
by Children’s Hospital of Philadelphia

Hemoglobin. Credit: CC0 Public Domain
The genetic blood disorders sickle cell disease and beta-thalassemia are caused by errors in the genes for hemoglobin, a protein in red blood cells that carries oxygen from the lungs to tissues throughout the body. In utero, the gamma-globin gene produces fetal hemoglobin, but after birth, this gene is switched off and the beta-globin gene is turned on, producing adult hemoglobin. Patients with sickle cell disease and beta-thalassemia have mutations in the beta-globin gene, which leads to mutant hemoglobin production and serious health complications, ranging from delayed growth to chronic pain and stroke.
Since the gamma-globin gene in these patients is not mutated and produces functional hemoglobin, there has been interest among researchers in reversing the switch from fetal to adult hemoglobin in these patients. Doing so would require identifying and exploiting factors responsible for silencing fetal hemoglobin and promoting adult hemoglobin production. Prior studies have identified BCL11A and LRF as transcription factors involved in silencing fetal-type globin genes HBG1 and HBG2, but further studies have shown that a complex array of transcriptional factors are involved in this process.
To identify which transcription factors are implicated, researchers from Children's Hospital of Philadelphia (CHOP), led by postdoctoral fellow Kunhua Qin, Ph.D. in collaboration with the laboratory of Junwei Shi, Ph.D., assistant professor of cancer biology at the University of Pennsylvania, performed a genetic screen using CRISPR-Cas9. Through this screen, they identified two members of the NFI transcription factor family—NFIA and NFIX—as HBG1 and HBG2 repressors. The researchers found that NFIA and NFIX are both elevated in adult red blood cells compared to fetal cells, and in cultured cells and mouse models, they found that that these transcription factors work together to repress fetal hemoglobin genes HBG1 and HBG2.
Using a combination of genomic profiling, genome editing, and DNA binding assays, the researchers found that NFIA and NFIX work in two ways to silence fetal hemoglobin: together they stimulate the expression of BCL11A, a known silencer of the HBG1/2 genes, and they also directly repress HBG1/2 genes. The findings were published today in Nature Genetics.
"Our study shows that transcription factors NFIA and NFIX have dual activity, both turning genes on and turning others off, the combined effect of which is the silencing of fetal hemoglobin," said senior study author Gerd A. Blobel, MD, Ph.D., an investigator and holder of the Frank E. Weise III Endowed Chair in Pediatric Hematology at Children's Hospital of Philadelphia. "This research demonstrates how deeply complex the transition from fetal to adult hemoglobin is, but by better understanding the factors involved, future research could focus on exploiting these transcription factors as therapeutic targets."
More information: Gerd Blobel, Dual function NFI factors control fetal hemoglobin silencing in adult erythroid cells, Nature Genetics (2022). DOI: 10.1038/s41588-022-01076-1. www.nature.com/articles/s41588-022-01076-1
Journal information: Nature Genetics
Provided by Children’s Hospital of Philadelphia





Post comments