
by Technion - Israel Institute of Technology
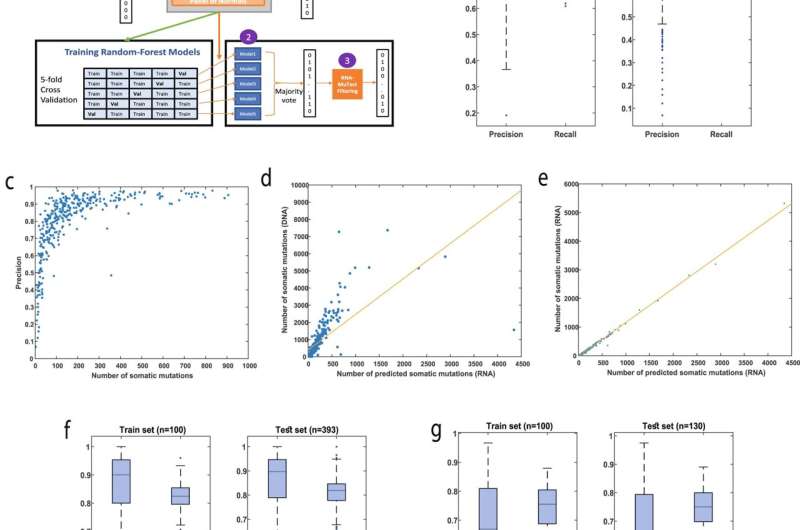
Summary of pipeline predictions. a An overview of the RNA-MuTect-WMN pipeline: In the training set (n = 100, green arrows), RNA-MuTect is applied on tumor RNA and a matched-normal DNA to obtain a list of variants labeled as somatic or germline. A random forest classifier is then trained with the collected set of features for each variant in a 5-fold cross validation manner. In the test set (orange arrows), 3 steps are performed: (1) MuTect is applied with tumor RNA and without a matched-normal sample, to yield a list of mixed somatic and germline variants. (2) The five trained models are then applied to this set of variants and classify them as either somatic or germline in a majority vote manner. (3) Finally, the predicted set of variants is further filtered by the RNA-MuTect filtering steps. b Distribution of precision and recall values on validation (left) and test (right) sets computed for each sample. Box plots show median, 25th, and 75th percentiles. The whiskers extend to the most extreme data points not considered outliers, and the outliers are represented as dots. c Precision as the function of the number of true somatic mutations per sample. d Correlation between the number of predicted somatic mutations and the number of somatic mutations as determined by DNA with a matched-normal DNA sample. e Correlation between the number of predicted somatic mutations and the number of somatic mutations as determined by RNA with a matched-normal DNA sample. f Distribution of precision and recall values on validation (left) and test (right) sets computed for each sample in the lung dataset. Box plots show median, 25th, and 75th percentiles. The whiskers extend to the most extreme data points not considered outliers, and the outliers are represented as dots. g Distribution of precision and recall values on validation (left) and test (right) sets computed for each sample in the colon dataset. Box plots show median, 25th, and 75th percentiles. The whiskers extend to the most extreme data points not considered outliers, and the outliers are represented as dots. Source data are provided as a Source Data file. Credit: Nature Communications (2022). DOI: 10.1038/s41467-022-30753-2
Can immunotherapy treatment help this cancer patient? And if it can, which specific treatment should be used? Oncologists routinely ask themselves these questions. Insurance companies also ask it because immunotherapy is expensive. Patients ask if this novel treatment can save their lives. Now, a new study by Professor Keren Yizhak, from the Ruth and Bruce Rappaport Faculty of Medicine at the Technion–Israel Institute of Technology, uses Artificial Intelligence to create a simple and inexpensive method of answering this question for each individual patient. Prof. Yitzhak's findings were recently published in Nature Communications.
Immunotherapy is a recent development in the world of cancer treatments. It has given full remission to patients who could not be helped by other means, and it reduces many of the side effects of chemotherapy. There are multiple immunotherapeutic treatments, but they differ from the new study since the principle under which they all operate is stimulating the patient's immune system to attack the tumor cells.
How does the immune system distinguish between the cancer cells it should attack and the healthy cells of the body? The more mutations the tumor has amassed, the more it differs from the "normal" cells, and thus immunotherapy can be more effective. This characteristic is called Tumor Mutation Burden (TMB). A higher TMB means more new mutations. Prof. Yizhak's method significantly simplifies measuring the TMB.
In order to measure the TMB the way it is done now, cells are taken from the tumor and their DNA is compared to DNA from the patient's healthy cells. Prof. Yizhak and her group propose two changes to this process.
The first change, already explored in a previously published article by the group, is comparing RNA molecules rather than DNA molecules. This makes a difference because DNA molecules contain the entirety of the human genome while RNA molecules are small parts of the genetic code, copied out to be used as instructions within the cell. In their previous study, the group showed that RNA molecules can also be used to identify the cancer-specific mutations.
The innovation in the group's most recent article is two-fold: first, the elimination of a need to compare the RNA from the tumor to DNA from healthy cells. As a result, a smaller amount of genetic material needs to be sequenced so the patient needs to be subjected to one less procedure. Instead of comparing the genetic material from the tumor to the patient's own healthy genetic material, Prof. Yizhak's team developed a machine-learning algorithm. The algorithm was trained to recognize aberrations from the healthy genome and to tell them apart from the natural variation that exists between people. Second, using these predictions, they were able to compute an RNA-based TMB metric. In fact, this method proved to be more effective than the standard method in estimating the predicted effectiveness of immunotherapy for a given patient. This is thought to be the case because the RNA contains the parts of the genome that are in constant use and can therefore initiate an immune response. Mutations in parts of the genome that are not in use are less likely to affect the cell's operation.
The development of the algorithm was made possible by using a large existing database of sequenced RNA from cancer patients, on which the algorithm could be trained. In fact, Prof. Yizhak's laboratory is a computational, "dry" lab. Computational labs make use of the large amounts of clinical data collected by the scientific community around the world, using it to achieve new discoveries and to develop new tools to assist patients. The study discussed here was led by Dr. Rotem Katzir and B.Sc. student Noam Rudberg, both from the Henry and Marilyn Taub Faculty of Computer Science.
More information: Rotem Katzir et al, Estimating tumor mutational burden from RNA-sequencing without a matched-normal sample, Nature Communications (2022). DOI: 10.1038/s41467-022-30753-2
Journal information: Nature Communications
Provided by Technion - Israel Institute of Technology






Post comments