
by Jelena Pupavac, Wellcome Trust Sanger Institute
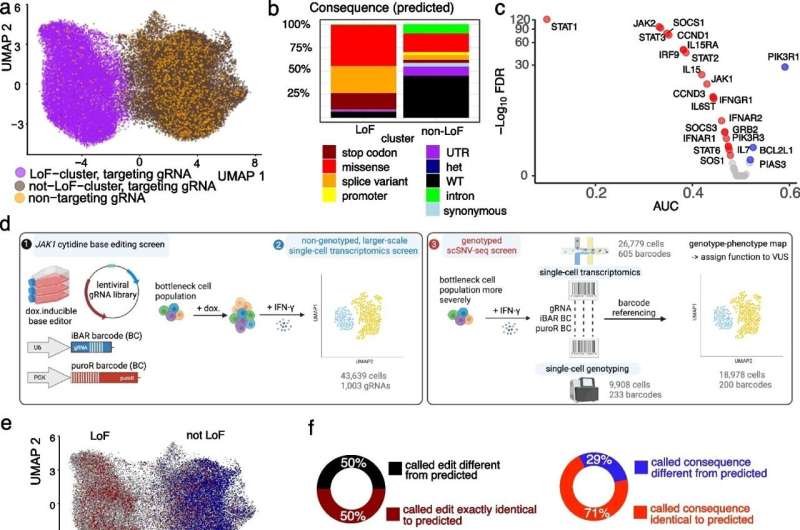
A single-cell base editor screen tiling across JAK1 is improved by coupling genotype with transcriptome. a UMAP of LoF and not LoF meta-clusters for the non-genotyped experiment including all cells with a uniquely assigned gRNA. NT-gRNAs are highlighted in orange. b Distribution of consequences of the predicted mutations for each cluster. c Differential gene expression analysis of JAK-STAT pathway genes between the LoF cluster and non-targeting gRNAs. AUC < 0.5 indicates downregulation (red, if significant) and AUC > 0.5 upregulation (blue, if significant). d Overview of high-throughput SNV phenotyping. Base editing of JAK1 was achieved through the introduction of a barcoded gRNA library into a doxycycline-inducible cytidine base editor expressing HT-29 cancer cells (left panel, 1). After editing, cells were induced with IFN-γ before single-cell transcriptomics (left panel, 2) or bottlenecked and processed for targeted single-cell DNA sequencing (right panel, 3). Transcriptomes and edited genotypes of single cells were linked through genetic barcodes to assign function to variants of unknown significance (VUS). e UMAP combining the non-genotyped (gray) data set with all genotyped cells with confidently called genotype (GT, 18,978 cells). Red and blue indicate edited and wild-type (WT) cells respectively. f Percentage of barcodes for which the called homozygous DNA editing is exactly the same as predicted based on complete editing in the window (maroon/black) or for which the functional consequences of the edit on the protein sequence are the same (red/blue). g UMAPs highlighting mutational consequences for the predicted genotypes (upper, non-genotyped data set) compared to the called genotypes (lower, genotyped data set). The colored cells are homozygous stop codon (brown), splice (yellow), or missense variants (red), with other cells shown in gray. Compare to Additional file 1: Fig. S2i. h Percentages of cells showing the consequence of mutations from actual genotyping in LoF and not LoF clusters. For the assignment of probable consequences using VEP, only homozygous mutations were included, as heterozygous edits are not expected to have a strong functional consequence. See also Additional file 1: Figs. S1 and S2. Credit: Genome Biology (2024). DOI: 10.1186/s13059-024-03169-y
Scientists have developed a new screening tool to uncover how genetic changes affect gene activity and can lead to diseases such as cancer, autoimmunity, neurodegeneration and cardiovascular disease. This new tool enables the investigation of thousands of DNA mutations identified by genetic studies in one experiment, guiding the development of advanced diagnostics and treatments.
The technique, called scSNV-seq, enables researchers to rapidly assess the impact of thousands of genetic changes in cells that have never been screened before, directly connecting these changes to how those same cells operate. This provides a comprehensive view from which researchers can pinpoint the mutations that contribute to disease. This will offer crucial insights for developing targeted therapies.
In the new study published in Genome Biology, researchers from the Wellcome Sanger Institute and their collaborators at Open Targets and EMBL's European Bioinformatics Institute (EMBL-EBI) applied scSNV-seq to the blood cancer gene JAK1.
The technique accurately assessed the impact of JAK1 mutations, revealing for the first time that certain mutations caused a "halfway house" phenotype cycling between different states. This is not possible under previous approaches.
The technique is designed to demonstrate versatility across cell types, including hard-to-culture primary cells like T cells and stem-cell-derived neurons, as well as various editing methods such as base editing and prime editing. Applied on a large scale, scSNV-seq could transform understanding of the genetic changes driving cancer and decoding genetic risk for Alzheimer's, arthritis, diabetes, and other complex diseases.
Advances in human genetics combined with the increasing affordability of DNA sequencing technologies have unveiled hundreds of thousands of disease-related genetic variants that are increasing at a staggering rate. Yet, tools to interpret them lag behind, sometimes relying on tedious manual processes.
When using advanced gene-editing tools to introduce defined genetic mutations, using current screening methods, it is difficult to distinguish between cells where the editing did not work and those where it successfully introduced a harmless change without affecting the cell's behavior.
Researchers from the Wellcome Sanger Institute and their collaborators set out to address this with a new screening technique, scSNV-seq, which directly couples the specific genetic information in the genotype of a cell to its gene activity. The team tested the effectiveness of scSNV-seq by altering specific DNA bases within the JAK1 gene, which is linked to inflammation and cancer, to study their effects on cell behavior.
They demonstrated scSNV-seq could accurately categorize different types of genetic changes into three categories: benign, causing loss of function, and altering function. They showed certain mutations caused an intermediate phenotype cycling between different states—an observation not possible under existing approaches.
Dr. Sarah Cooper, first author of the study at the Wellcome Sanger Institute, said, "In an era where the rate of genetic variant discovery outpaces our ability to interpret their effects, scSNV-seq fills a major gap for studying challenging cells like T cells and neurons. We are already using it to shed light on the impact of Alzheimer's and Parkinson's risk variants on brain cells."
Dr. Andrew Bassett, senior author of the study at the Wellcome Sanger Institute, said, "Our technique is able to directly connect effects of mutations to how a cell behaves, revealing downstream impacts that previous technologies alone cannot deliver. The technique speeds up the identification of causal genetic mutations, which will allow better diagnosis and deepens our molecular understanding of diseases, paving the way for more targeted and effective treatments."
More information: Sarah E. Cooper et al, scSNV-seq: high-throughput phenotyping of single nucleotide variants by coupled single-cell genotyping and transcriptomics, Genome Biology (2024). DOI: 10.1186/s13059-024-03169-y
Provided by Wellcome Trust Sanger Institute






Post comments