
by University of Sussex
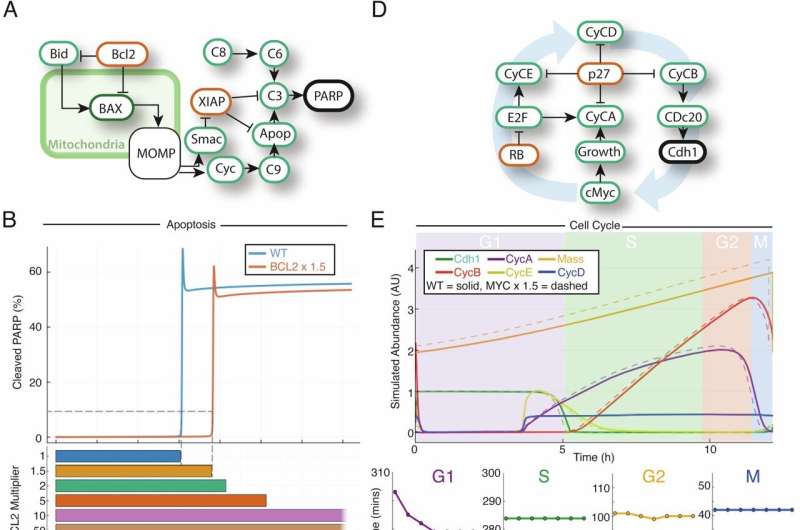
Computational modeling of the cell cycle and apoptosis reveal limited impact of archetypal double hit mutations on their respective molecular networks. Credit: Blood Cancer Journal (2024). DOI: 10.1038/s41408-024-01090-y
A study led by researchers at Brighton and Sussex Medical School (BSMS) has unveiled an approach to predicting the effectiveness of treatments for patients with diffuse large B-cell lymphoma (DLBCL), a common form of blood cancer.
The researchers utilized genomic sequencing data to create personalized simulations of individual patients that can quantify the impact of genetic mutations on cancer cell behavior. This innovative method promises to pave the way for personalized medicine, which will revolutionize clinical decision-making and advance care for heterogeneous blood cancers.
The research team, led by Dr. Simon Mitchell, Reader in Cancer Systems Biology at BSMS, focused on leveraging genomic data from DLBCL patients to simulate how specific mutations affect anti-apoptotic and pro-proliferative signaling within cancer cells. Unlike traditional approaches that rely on mutational clustering, the simulations provided a more comprehensive understanding of the interplay between multiple mutations. Rather than grouping patients sharing common mutations, their approach groups patients based on the impact of mutations within complex signaling networks.
The study found that personalized simulations successfully identified patients with varying prognoses (dismal, intermediate and good) across multiple datasets. This was achieved using data from whole-exome sequencing (WES) or targeted sequencing panels, providing robust predictions despite the mutational heterogeneity.
Unlike many statistical approaches, the predictive accuracy of the simulations improved with larger validation datasets, emphasizing the importance of integrating molecular network knowledge into the analysis. The models were particularly effective at pinpointing patients with co-occurring mutations that promote both cancer cell proliferation and resistance to apoptosis—and found patients that would be missed by traditional clustering methods.
Commenting on the study, Dr. Mitchell said, "This study supports the integration of genetic sequencing at the diagnosis stage of DLBCL to better determine patient prognosis. As sequencing costs decrease, we hope that this approach will become a standard diagnostic practice, enabling precise identification of patients who might benefit from alternative treatments.
"This study marks a significant step forward in the quest for personalized cancer treatment. By harnessing the power of computational modeling to place genomic data into context, we hope to pave the way for more accurate prognostic predictions and tailored therapeutic strategies. We believe such approaches promise a new era of precision medicine for blood cancer patients and potentially many others."
Beyond DLBCL, the computational modeling techniques demonstrated in this study have the potential to be applied to other types of cancer, particularly those characterized by high genetic heterogeneity.
As genomic data becomes more widely available and computational methods continue to evolve, these personalized simulations could play a critical role in the era of precision medicine, tailoring treatments to individual genetic profiles for better patient outcomes.
The study has been published in Blood Cancer Journal.
More information: Richard Norris et al, Patient-specific computational models predict prognosis in B cell lymphoma by quantifying pro-proliferative and anti-apoptotic signatures from genetic sequencing data, Blood Cancer Journal (2024). DOI: 10.1038/s41408-024-01090-y
Journal information: Blood Cancer Journal
Provided by University of Sussex







Post comments