
by UT Southwestern Medical Center
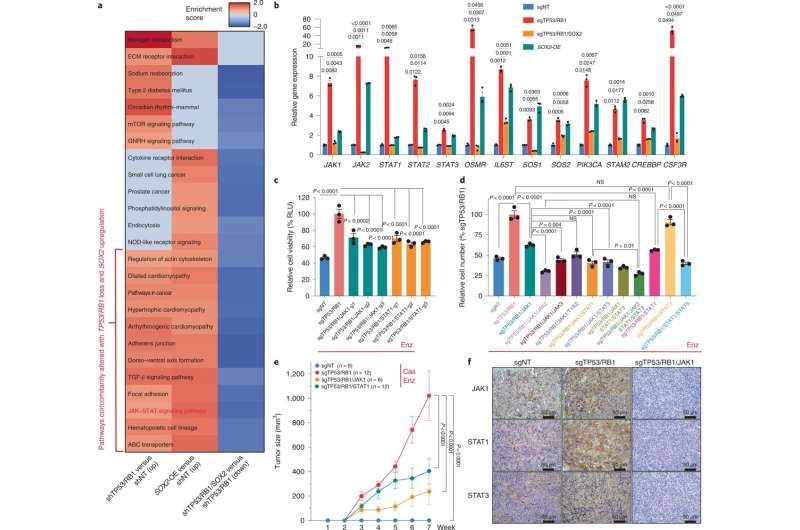
JAK–STAT signaling is required for Enz resistance in TP53/RB1-deficient mCRPC. a, Heat map representing the significantly changed signaling pathways in LNCaP/AR cell lines transduced with annotated shRNAs based on GSEA analysis. Three comparisons are presented. Reads from n = 3 independently treated cell cultures in each group were used for analysis. Signaling pathways concomitantly altered with TP53/RB1 loss and SOX2 upregulation are labeled with a red bracket. b, Relative gene expression of canonical genes activated in the JAK–STAT signaling pathway in LNCaP/AR cells transduced with Cas9 and annotated guide RNAs; n = 3 independently treated cell cultures. P values were calculated using a two-way ANOVA with a Bonferroni multiple-comparison test. c, Relative cell numbers of LNCaP/AR cells transduced with Cas9 and annotated CRISPR guide RNAs. Cells were treated with 10 µM Enz for 8 d, and cell numbers (viability) were measured using a CellTiter-Glo assay, with all values normalized to the sgTP53/RB1 group; n = 3 independently treated cell cultures. P values were calculated by one-way ANOVA with a Bonferroni multiple-comparison test; RLU, relative light units. d, Relative cell numbers of LNCaP/AR cells transduced with Cas9 and annotated CRISPR guide RNAs. Cells were treated with 10 µM Enz for 8 d, and cell numbers (viability) were measured using a CellTiter-Glo assay, with all values normalized to the sgTP53/RB1 group; n = 3 independently treated cell cultures. P values were calculated by one-way ANOVA with a Bonferroni multiple-comparison test; NS, not significant. e, Tumor growth curve of xenografted LNCaP/AR cells transduced with Cas9 and annotated guide RNAs in castrated mice. Cas denotes castration 2 weeks before grafting. Enz denotes Enz treatment at 10 mg kg–1 from day 1 of grafting; n = number of independent xenografted tumors in each group (two tumors per mouse); sgNT, n = 8 tumors; sgTP53/RB1, n = 12 tumors; sgTP53/RB1/JAK1, n = 8 tumors; sgTP53/RB1/STAT1, n = 12 tumors. P values were calculated by two-way ANOVA with a Bonferroni multiple-comparison test. f, IHC staining of JAK–STAT proteins on annotated xenografted tumor slides showing representative images of n = 2 independent tumors. Credit: Nature Cancer (2022). DOI: 10.1038/s43018-022-00431-9
Stopping prostate cancer cells from morphing into other cell types can overcome the resistance they develop to widely prescribed therapies, a study led by UT Southwestern scientists suggests. The findings, published in Nature Cancer, could lead to a new approach to fight this deadly disease, the second-most common cancer in American men that kills nearly 35,000 annually in the U.S.
"We think this novel combination therapy could dramatically improve clinical outcomes of prostate cancer patients and hopefully save many lives," said study leader Ping Mu, Ph.D., Assistant Professor of Molecular Biology and a member of the Harold C. Simmons Comprehensive Cancer Center.
Drugs that target the androgen receptor (AR), a key protein for prostate development and maintenance, have revolutionized prostate cancer management in recent decades, extending the lives of hundreds of thousands of patients. However, explained Dr. Mu, these therapies fail over time as prostate tumors develop resistance to them.
In the past five years, Dr. Mu and other cancer researchers discovered that one reason these tumors become resistant is through a phenomenon called lineage plasticity, in which malignant prostate cells revert to an earlier stage in development and take on a new identity, becoming a different cell type that no longer depends on the AR. Although lineage plasticity has been discovered in other cancer types, including lung cancer, breast cancer, and melanoma, the molecular mechanism behind it has remained largely unknown, stymieing efforts to develop therapies to prevent resistance from developing.
To better understand what drives lineage plasticity and resistance, Dr. Mu and his colleagues compared cells that were resistant and sensitive to AR-targeting drugs using multiple lab models: human prostate cancer cell lines growing in petri dishes, three-dimensional "organoids" made of human and mouse prostate cancer cells that mimic the structure of natural prostate cancers, and mouse models of prostate cancer. Using different analytical methods including single-cell RNA sequencing, the researchers searched for key molecular pathways that separated the resistant cells from the sensitive ones.
Their search revealed that a particular signaling pathway called Janus kinase-signal transducer and activator of transcription (JAK-STAT) appeared to drive both lineage plasticity and resistance. When the researchers used a genetic technique to separately knock out the 11 major genes that make up this pathway, they discovered that genes known as JAK1 and STAT1 played key roles in these phenomena. Eliminating these genes caused cancerous prostate cells that had shifted into new cell types to revert to their original identities and become sensitive to current therapy again.
Treating cancer cells with drugs that inhibit these genes had a similar effect, Dr. Mu explained, not only ending their lineage plasticity but also re-sensitizing them to AR-targeting therapies. When resistant prostate cancer cells were treated with both JAK1 and STAT1 inhibitors along with an AR-targeting drug, these cancer cells lost their ability to divide and survive.
Using a similar strategy could offer a new way to overcome resistance in human prostate cancer patients, Dr. Mu said, an approach he and his colleagues plan to eventually test in a clinical trial.
More information: Su Deng et al, Ectopic JAK–STAT activation enables the transition to a stem-like and multilineage state conferring AR-targeted therapy resistance, Nature Cancer (2022). DOI: 10.1038/s43018-022-00431-9
Journal information: Nature Cancer
Provided by UT Southwestern Medical Center







Post comments