
by Ian Demsky, Memorial Sloan Kettering Cancer Center
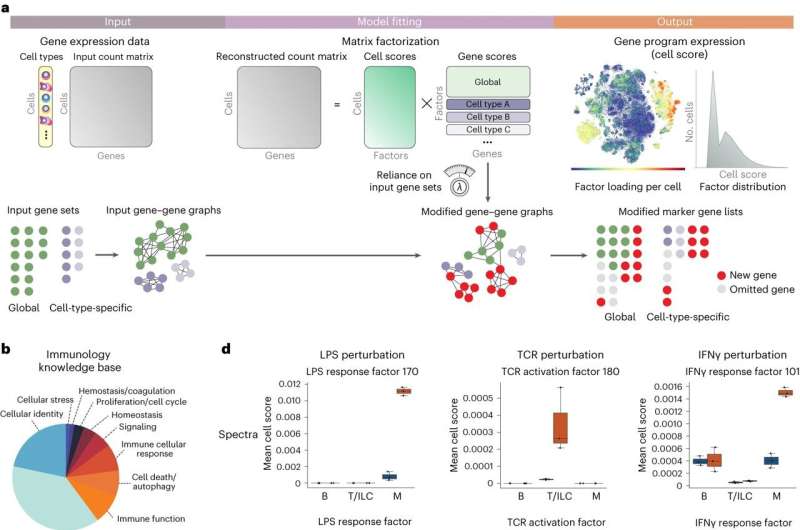
Spectra uses gene sets and cell types to guide gene program discovery from scRNA-seq data. a, As input, Spectra receives a gene expression count matrix with cell-type labels for each cell as well as predefined gene sets, which it converts to a gene–gene graph. The algorithm fits a factor analysis model using a loss function that optimizes reconstruction of the count matrix and guides factors to support the input gene–gene graph. As output, Spectra provides factor loadings (cell scores) and gene programs corresponding to cell types and cellular processes (factors). b, Gene set categories in the immunology knowledge base. c, Design of the perturbation experiments from Kartha et al.8. PBMCs (n = 23,754) from healthy human donors (n = 3) were incubated for 6 h with LPS, PMA or recombinant human IFNγ. d, Ability of different algorithms to identify gene programs associated with biological perturbations in the PBMC dataset. For select factors, mean per-donor cell scores are provided for T cells or innate lymphoid cells (T/ILCs), B cells (B) and myeloid cells (M; n = 3 donors). Boxes and lines represent interquartile range (IQR) and median, respectively; whiskers represent 1.5× IQR. Credit: Nature Biotechnology (2023). DOI: 10.1038/s41587-023-01940-3
Researchers at Memorial Sloan Kettering Cancer Center (MSK) have developed a new open-source computational method, dubbed Spectra, which improves the analysis of single-cell transcriptomic data.
By guiding data analysis in a unique way, Spectra can offer new insights into the complex interplay between cells—like the interactions between cancer cells and immune cells, which are critical to improving immunotherapy treatments.
The team's approach and findings were recently published in Nature Biotechnology.
Spectra, the researchers note, can cut through technical "noise" to identify functionally relevant gene expression programs, including those that are novel or highly specific to a particular biological context.
The algorithm is well suited to study data from large patient cohorts and to suss out clinically meaningful patient characteristics, the MSK team writes in a research briefing that accompanies the study, adding that Spectra is ideal for identifying biomarkers and drug targets in the burgeoning field of immuno-oncology.
Additionally, the MSK team has made Spectra freely available to researchers around the world.
"I'm trained as a computer scientist," says study senior author Dana Pe'er, Ph.D., who chairs the Computational and Systems Biology Program at MSK's Sloan Kettering Institute. "Every single tool I build, I strive to make robust so it can be used in many contexts, not just one. I also try and make them as accessible as possible."
"I'm happy to discover new biology," she continues. "And I'm just as happy—perhaps happier—to build a foundational tool that can be used by the wider community to make many biological discoveries."
Along with researchers at MSK, teams from several institutions are already using Spectra to study a variety of diseases, Dr. Pe'er adds.
The single-cell revolution
Over the past decade, the "single-cell revolution" has transformed human understanding of health and disease. Single-cell technologies allow scientists to study the individual cells in a tissue sample or set of samples—a tumor, for example—and see not only the variety of cell types that are present (such as cancer cells versus various types of immune cells) but also which genes are active in each cell, shedding new light on cell states and cell interactions.
The technology has fostered new understandings about how cells adapt and respond to changing conditions in health and disease—including the development of resistance to cancer treatments.
The problem is that the mind-boggling amount of data generated by single-cell methods is challenging to sift through and correctly interpret. This is particularly true when trying to look at gene programs—genes that work together to accomplish a particular task —that are active across multiple cell types in a tissue, Dr. Pe'er explains.
"This is especially important for studying the interactions between cancer cells and immune cells, which involve highly overlapping gene programs," she says. "This causes some serious statistical problems that can lead to incredibly misleading results."
The team Dr. Pe'er assembled—led by co-first authors Russell Kunes, a doctoral student trained in statistics, and Thomas Walle, MD, a physician-scientist with expertise in immuno-oncology—not only developed the improved method for guiding the data analysis, but they also created a user-friendly interface to facilitate its adoption by other scientists.
"Our desire to develop an improved method brought together researchers with vastly different expertise in statistics, computational biology, and immunology," Dr. Walle writes in the research briefing. "Spectra was a journey of mutual learning, with the shared goal of making complex biology explainable."
Guiding the data analysis
For the paper, the researchers applied Spectra across two breast cancer immunotherapy data sets and a lung cancer atlas, together totaling more than 1.5 million cells from 375 individuals in 21 studies, demonstrating Spectra's ability to overcome the limitations of traditional analyses at scale.
Spectra gets its power by guiding the data analysis with a body of existing scientific knowledge—libraries of gene programs generated from previous data by experts in their fields. And while this starting knowledge can directly guide single-cell data analysis, the program also adapts to the data at hand, helping to identify new and modified gene programs. (This adaptive property allowed the scientists to uncover a novel cancer invasion program in tumor-associated macrophages related to anti-PD-1 immunotherapy resistance, they note in the paper.)
Spectra's unique design also takes into account information about the genes that define different cell types, making it more adept at finding gene programs that underlie cellular functions, as opposed to cellular identity.
"Spectra, for example, allows us to separate T cells that are exhausted from the tumor-reactive T cells, which are actively fighting a person's cancer," Dr. Pe'er says. "And it helps us to see the differences in gene activation between the two—which is quite challenging to unravel in complex contexts like the tumor microenvironment."
Additionally, the authors note, the ability to transfer gleanings from one data set directly to another will speed up and streamline discovery, allowing researchers to refine knowledge across single-cell sequencing studies without requiring complex data integration.
More information: Russell Z. Kunes et al, Supervised discovery of interpretable gene programs from single-cell data, Nature Biotechnology (2023). DOI: 10.1038/s41587-023-01940-3
Decoding the building blocks of cellular processes from single-cell transcriptomics data, Nature Biotechnology (2023). DOI: 10.1038/s41587-023-01967-6
Github: github.com/dpeerlab/spectra
Journal information: Nature Biotechnology
Provided by Memorial Sloan Kettering Cancer Center






Post comments