
by Bendta Schroeder, Massachusetts Institute of Technology
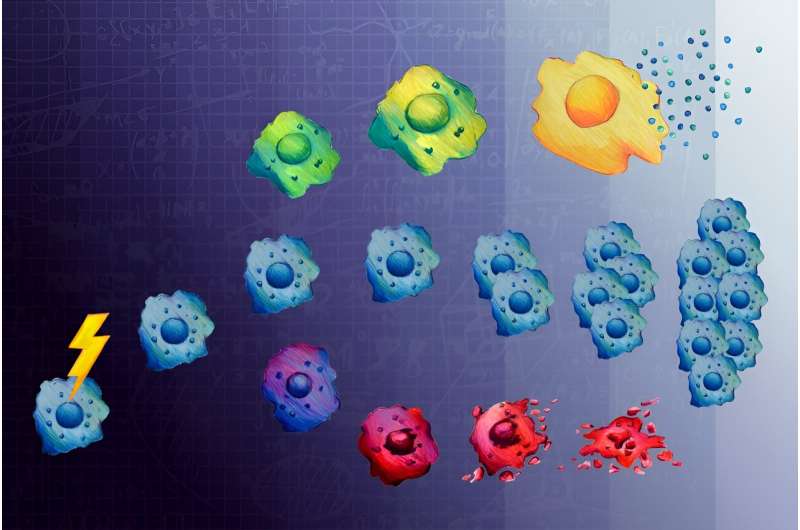
Normal cells (blue) entering senescence (green to yellow) or apoptosis (purple to red) after treatment with chemotherapy. Credit: Bendta Schroeder
Despite the proliferation of novel therapies such as immunotherapy or targeted therapies, radiation and chemotherapy remain the frontline treatment for cancer patients. About half of all patients still receive radiation and 60%–80% receive chemotherapy.
Both radiation and chemotherapy work by damaging DNA, taking advantage of a vulnerability specific to cancer cells. Healthy cells are more likely to survive radiation and chemotherapy since their mechanisms for identifying and repairing DNA damage are intact. In cancer cells, these repair mechanisms are compromised by mutations. When cancer cells cannot adequately respond to the DNA damage caused by radiation and chemotherapy, ideally, they undergo apoptosis or die by other means.
However, there is another fate for cells after DNA damage: senescence—a state where cells survive, but stop dividing. Senescent cells' DNA has not been damaged enough to induce apoptosis but is too damaged to support cell division. While senescent cancer cells themselves are unable to proliferate and spread, they are bad actors in the fight against cancer because they seem to enable other cancer cells to develop more aggressively.
Although a cancer cell's fate is not apparent until a few days after treatment, the decision to survive, die, or enter senescence is made much earlier. But, precisely when and how that decision is made has not been well understood.
In an open-access study of ovarian and osteosarcoma cancer cells that was published July 19 in Cell Systems, MIT researchers show that cell signaling proteins commonly associated with cell proliferation and apoptosis instead commit cancer cells to senescence within 12 hours of treatment with low doses of certain kinds of chemotherapy.
"When it comes to treating cancer, this study underscores that it's important not to think too linearly about cell signaling," says Michael Yaffe, who is a David H. Koch Professor of Science at MIT, the director of the MIT Center for Precision Cancer Medicine, a member of MIT's Koch Institute for Integrative Cancer Research, and the senior author of the study. "If you assume that a particular treatment will always affect cancer cell signaling in the same way—you may be setting yourself up for many surprises, and treating cancers with the wrong combination of drugs."
Using a combination of experiments with cancer cells and computational modeling, the team investigated the cell signaling mechanisms that prompt cancer cells to enter senescence after treatment with a commonly used anti-cancer agent. Their efforts singled out two protein kinases and a component of the AP-1 transcription factor complex as highly associated with the induction of senescence after DNA damage, despite the well-established roles for all of these molecules in promoting cell proliferation in cancer.
The researchers treated cancer cells with low and high doses of doxorubicin, a chemotherapy that interferes with the function with topoisomerase II, an enzyme that breaks and then repairs DNA strands during replication to fix tangles and other topological problems.
By measuring the effects of DNA damage on single cells at several time points ranging from six hours to four days after the initial exposure, the team created two datasets. In one dataset, the researchers tracked cell fate over time. For the second set, researchers measured relative cell signaling activity levels across a variety of proteins associated with responses to DNA damage or cellular stress, determination of cell fate, and progress through cell growth and division.
The two datasets were used to build a computational model that identifies correlations between time, dosage, signal, and cell fate. The model identified the activities of the MAP kinases Erk and JNK, and the transcription factor c-Jun as key components of the AP-1 protein likewise understood to involved in the induction of senescence. The researchers then validated these computational findings by showing that inhibition of JNK and Erk after DNA damage successfully prevented cells from entering senescence.
The researchers leveraged JNK and Erk inhibition to pinpoint exactly when cells made the decision to enter senescence. Surprisingly, they found that the decision to enter senescence was made within 12 hours of DNA damage, even though it took days to actually see the senescent cells accumulate. The team also found that with the passage of more time, these MAP kinases took on a different function: promoting the secretion of proinflammatory proteins called cytokines that are responsible for making other cancer cells proliferate and develop resistance to chemotherapy.
"Proteins like cytokines encourage 'bad behavior' in neighboring tumor cells that lead to more aggressive cancer progression," says Tatiana Netterfield, a graduate student in the Yaffe lab and the lead author of the study. "Because of this, it is thought that senescent cells that stay near the tumor for long periods of time are detrimental to treating cancer."
This study's findings apply to cancer cells treated with a commonly used type of chemotherapy that stalls DNA replication after repair. But more broadly, the study emphasizes that "when treating cancer, it's extremely important to understand the molecular characteristics of cancer cells and the contextual factors such as time and dosing that determine cell fate," explains Netterfield.
The study, however, has more immediate implications for treatments that are already in use. One class of Erk inhibitors, MEK inhibitors, are used in the clinic with the expectation that they will curb cancer growth.
"We must be cautious about administering MEK inhibitors together with chemotherapies," says Yaffe. "The combination may have the unintended effect of driving cells into proliferation, rather than senescence."
In future work, the team will perform studies to understand how and why individual cells choose to proliferate instead of enter senescence. Additionally, the team is employing next-generation sequencing to understand which genes c-Jun is regulating in order to push cells toward senescence.
More information: Tatiana S. Netterfield et al, Biphasic JNK-Erk signaling separates the induction and maintenance of cell senescence after DNA damage induced by topoisomerase II inhibition, Cell Systems (2023). DOI: 10.1016/j.cels.2023.06.005
Journal information: Cell Systems
Provided by Massachusetts Institute of Technology
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.





Post comments