
by Ari Navetta, Broad Institute of MIT and Harvard
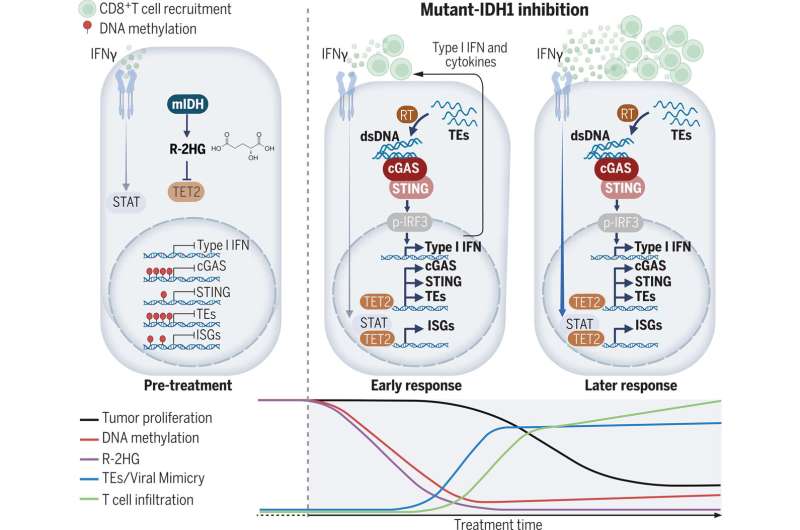
mIDH1 inhibition restores immune surveillance. Credit: Science (2024). DOI: 10.1126/science.adl6173
A number of cancers, including certain brain and liver tumors, as well as myeloid leukemias, are driven by mutations in the enzyme isocitrate dehydrogenase 1, or IDH1. Drugs that block mutated IDH1 (mIDH1) have recently come onto the market and effectively slow tumor growth, but the exact mechanism at work to stall tumors is still unclear.
Reporting in Science, researchers at Massachusetts General Hospital and the Broad Institute of MIT and Harvard have made a surprising discovery about these drugs.
They found that mIDH1 inhibitors trick the tumor cells into thinking they are infected with a virus, causing the immune system to mount an antiviral response. This mechanism, known as "viral mimicry," relies on the relics of ancient viral infections that remain scattered throughout the genome but are usually unexpressed. Treatment with mIDH1 blockers unearths these remnants, triggering an immune response against the tumor cells.
These findings stem from an observation first made several years ago in the lab of study co-senior author Nabeel Bardeesy, an associate member of the Cancer Program at Broad and a cancer geneticist at MGH.
"We felt that the gap in understanding biological effects of mIDH1 inhibitors was a major challenge in the field, limiting our ability to most effectively use these drugs," Bardeesy said.
After generating the first-ever mouse model of IDH1-mutated liver cancer, Bardeesy and his colleagues realized that only mice with a functioning immune system responded to mIDH1 inhibitors. To learn why, the Bardeesy lab has been collaborating with co-senior author Robert Manguso and co-first authors Meng-Ju Wu, Hiroshi Kondo, and Ashwin Kammula.
"These findings reveal a completely unexpected and fascinating mechanism that explains the efficacy of the drug," Manguso, who is a principal investigator at MGH, a co-director of the Tumor Immunology Discovery Engine (TIDE) group, and a Cancer Program associate member at Broad, said.
IDH1 normally facilitates the activity of enzymes called demethylases, which remove chemical flags called methylation marks from DNA, allowing genes to be transcribed into RNA. In its mutant form, however, IDH1 instead creates a metabolite called 2-hydroxyglutarate (2HG) that directly interferes with demethylases, causing the tumor cell to actively suppress expression of a wide range of genes.
By preventing the production of 2HG, mIDH1 inhibitors restore demethylases' activity, leading to the re-expression of many genes, including those that code for the ancient viral enzymes, which alert the immune system to mobilize an antiviral response.
This immune response relies on the expression of a gene called cGAS, which produces part of a protein that detects retroviruses. In cells with mutant IDH1, this gene is silenced due to the buildup of 2HG, but mIDH1 inhibitors allow demethylases to facilitate cGAS expression.
cGAS detects retroviruses and sets in motion a series of cellular events culminating in the production of interferon and other cytokines that trigger an antiviral immune response. This suggests that silencing the cGAS pathway is one method IDH1 mutant tumors use to evade immune detection.
"Viral mimicry triggers an immune response and enhances cancer cell killing using immune cells," co-first author Wu said. "This mechanism could be applied to other cancers with similar metabolic or epigenetic alterations."
The study's findings suggest that coming up with ways to harness the effects of viral mimicry could lead to more effective cancer treatment using mIDH1 inhibitors.
"Elucidating these mechanisms changes the way we think about combining mIDH1 inhibitors with other medicines and suggests possible paths tumors take to drug resistance," Bardeesy said.
More information: Meng-Ju Wu et al, Mutant IDH1 inhibition induces dsDNA sensing to activate tumor immunity, Science (2024). DOI: 10.1126/science.adl6173
Journal information: Science
Provided by Broad Institute of MIT and Harvard





Post comments