
by Ana María Rodríguez, Ph.d., Baylor College of Medicine
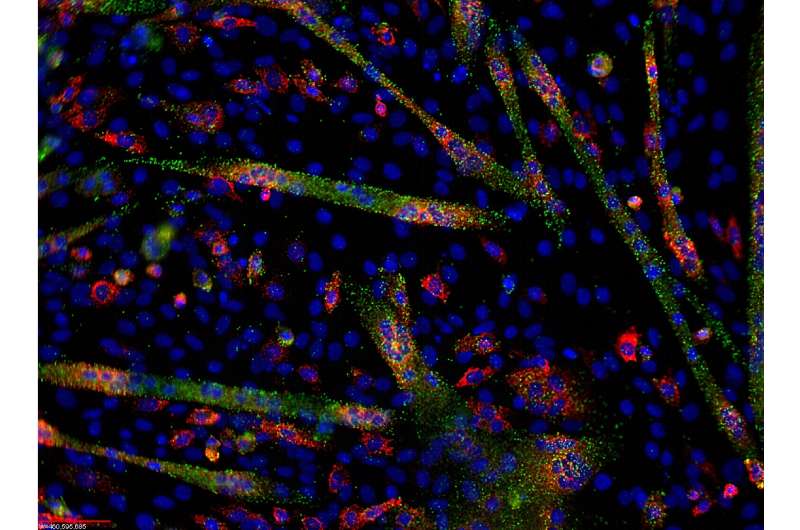
MicroRNA and mRNA visualization in differentiating C1C12 cells. Credit: Ryan Jeffs/Wikipedia
What started as Ninad Oak's side project turned out into something much larger, his doctorate thesis.
"The project started as my qualifying exam that I proposed at the end of my first year of graduate school," said Oak, a graduate student in molecular and human genetics in Dr. Sharon E. Plon's lab. "This was an off topic qualifying exam at the time, meaning the lab had not worked on this topic before."
One of the main interests of the Plon lab is cancer predisposition, in particular looking at protein coding regions of gene variants that may be responsible for susceptibility to childhood cancers.
"I started the project thinking that we had focused on protein coding regions for a long time. But they only represent 1 percent of the genome, so I thought that by looking at the remaining 99 percent we might find variations we have been missing that might explain some undiagnosed patient cases," Oak said.
In his qualifying exam, Oak proposed to look at variations on microRNA.
"Although the amount of microRNA that is found in the cell is often studied in human disease, microRNA variations that are associated with disease are understudied," Oak said.
MicroRNAs are small non-coding RNAs that are only about 18 to 25 nucleotides long; they are much smaller than genes that code for proteins, which are thousands of nucleotides long. MicroRNAs upregulate or downregulate the expression of more than 60 percent of genes by binding to matching sequences in other's genes' RNA. One microRNA might change the level of expression of up to 200 genes at a time, therefore affecting a number of different pathways.
Disturbing the normal function of microRNAs can lead to altered expression of their target genes, and this has been associated with a wide variety of human diseases, such as cancer, cardiovascular and developmental diseases.
"When he presented this proposal, I thought it was a good idea," said Plon, who is professor of pediatrics—oncology and molecular and human genetics at Baylor and director of the Cancer Genetics Clinical and Research Programs at Texas Children's Hospital.
ADmiRE helps prioritize microRNA variation linked to disease
Oak developed a novel computational tool called ADmiRE, which stands for Annotative Database for miRNA Elements. ADmiRE extensively annotates human microRNA variants to determine which ones are likely to contribute to or cause diseases.
"There were multiple challenges when I started working on this project," Oak said. "Most datasets of genomic sequencing are of whole exome sequencing (WES), which captures only protein coding regions. So first, I looked at how well WES datasets captured microRNAs and found that they captured about 50 percent."
The second challenge was to determine how well were microRNA mutations annotated by different annotation tools that already were available. These annotation tools allow researchers to add notes of explanation or comments that provide more information about each microRNA.
"There are various annotation tools that identify where a mutation is in general in the genome, not exclusively in microRNA. I found that these tools didn't annotate microRNA accurately; they tended to favor the potential change to a protein coding gene and not the impact on microRNAs. These tools also didn't include comprehensive information that would help us interpret and prioritize the potential role of that microRNA in disease," Oak said.
Oak worked on a microRNA annotation tool that would correctly annotate all microRNA variants and then used it to analyze one of the largest publicly available WES datasets of adults (gnomAD) to establish a baseline of microRNA variation in normal human populations.
"This approach allowed us to draw conclusions about how frequently microRNAs are variable in normal datasets," Oak said. "Knowing the background variation would help us identify potential microRNA variants in disease states."
To identify microRNAs that could be associated with disease, Oak developed a metric called allele frequency percentile score that shows how frequently a microRNA varies when compared to other microRNAs in these large datasets. He then selected a group of microRNAs that were in the lower quartile, essentially highlighting those with little variation. The reasoning is that microRNAs that are highly conserved in the adult population are so because otherwise disease may follow. Those highly conserved microRNAs would be candidates for being associated with disease.
The researchers then applied this new tool to analyze mutations across 10,000 cancer cases that included 32 cancer types in the Cancer Genome Atlas PanCancerAtlas WES dataset.
"We found miR-142 mutations linked to hematologic cancers, confirming the finding made a few years ago. Also, we found microRNA mutations in miR-21, which had not been previously associated with cancer. Our analysis with ADmiRE suggests that these mutations may contribute to mechanisms involved in esophageal cancer.
"At a personal level, I found this work very satisfying because I think it contributes a new technique to our lab that fills a gap in the field," Oak said. "From the scientific point of view, ADmiRE offers a new resource for researchers who have not found a genetic cause for a disease in protein coding genes. We have made this tool publicly available, and researchers can apply it to determine whether there is a signal in miRNA sequences. Maybe down the line this tool could be used by clinical laboratories."
"I think it is an important tool," Plon said. "Mutations in microRNA have been missed for many years, but I think ADmiRE will now allow labs that have mutation data to see if these mutations that we know are important play a role in the biology of human health."
More information: Ninad Oak et al, Framework for microRNA variant annotation and prioritization using human population and disease datasets, Human Mutation . DOI: 10.1002/humu.23668
Journal information: Human Mutation
Provided by Baylor College of Medicine





Post comments