
by Quinn Eastman, Emory University
Graphical abstract. Credit: Cell Reports (2024). DOI: 10.1016/j.celrep.2023.113662
A new Cell Reports paper from Bing Yao's lab in Emory's Department of Human Genetics provides insights into mechanisms underlying several neurodegenerative diseases, such as ALS (amyotrophic lateral sclerosis) and Alzheimer's.
It can be summarized in one line: TDP-43 keeps genetic zombies at bay.
Yao says that the zombies are just a part of the story, but they represent a window into what the protein TDP-43 does in our cells, and what happens when its function lapses and cells' regulatory machinery breaks down.
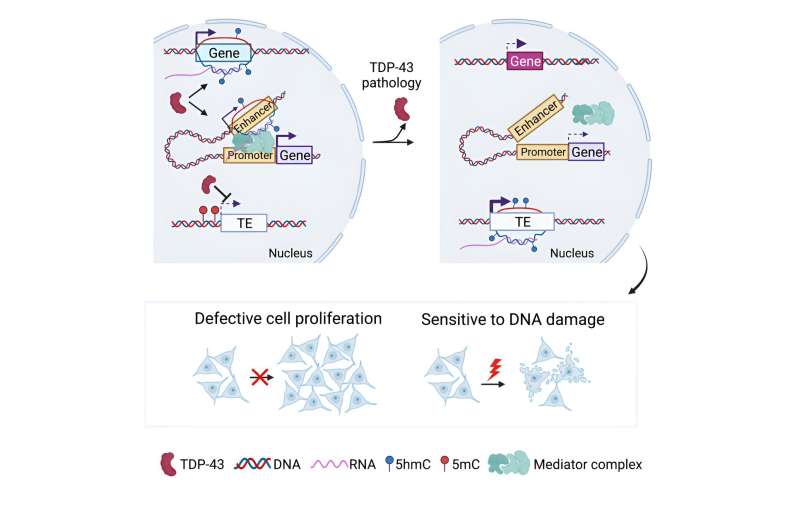
TDP-43 is one of those notorious aggregation-prone proteins familiar to researchers studying neurodegenerative diseases. It's like amyloid and tau, but perhaps not as famous. TDP-43's cytoplasmic aggregates are considered "the pathological hallmark" of ALS, and its aggregates also can be found in Alzheimer's brain samples.
"Most studies of TDP-43's function focus on it as an RNA-binding protein," Yao says. "But our analysis really captures how it is playing a genome-wide and epigenetic role, which is not fully appreciated."
Yao and his colleagues wanted to examine what happens when TDP-43 is clumping together in the cytoplasm, and thus is not doing what it's supposed to in the nucleus. So they created cell lines that have the TDP-43 gene chronically turned down. The cells divide more slowly and are more sensitive to DNA damage, but they're still alive.
A detailed analysis of what's wrong shows how critical TDP-43 is, and how it might deserve the name "guardian of the genome" just as much as the tumor suppressor p53.
Now we get to the zombies. In this case, the zombies are transposable elements, the occasionally mobile retrovirus-like sequences that make up a large part of our genomes.
Transposable elements (TEs) are usually inactive and not expressed, but if they become activated, several bad things can happen to cells, like DNA damage, inflammation and senescence. So cells have ways to prevent TEs from taking over.
The authors' analysis shows that in the absence of TDP-43, a large set of TEs get turned on. While other labs have shown that unsettling things happen to TEs without TDP-43, the elements themselves are difficult to analyze because their sequences are so similar to each other. By looking at the genomic context, this paper was able to assess which TE families are activated.
Yao and colleagues also analyze the effects of TDP-43 depletion with respect to the entire genome. TDP-43 normally maintains R-loops (three-stranded RNA-DNA structures in the nucleus) and facilitates their processing. Without TDP-43, the landscape of R-loops shifts. Some genes display more of them and some display less.
The patterns of DNA hydroxymethylation—an epigenetic modification associated with active genes—shift, too. In addition, the absence of TDP-43 appears to interfere with long-range interactions between enhancers and promoters, which are essential for proper gene expression.
"Our findings suggest that besides TDP-43, other RNA-binding proteins could have this kind of genome-wide role," Yao says.
In terms of next steps, Yao's team plans to analyze post-mortem tissue samples from Alzheimer's and ALS patients—because it's important to confirm that the same mechanism is operating in human diseases.
For therapeutic angles, some inconclusive clinical trials have tested anti-retrovirals in ALS, but Yao says it may be better to go after specific R-loops. He is crafting a strategy pairing CRISPR/dCas with an enzyme that targets RNA/DNA hybrid structures (RNAse H).
More information: Yingzi Hou et al, TDP-43 chronic deficiency leads to dysregulation of transposable elements and gene expression by affecting R-loop and 5hmC crosstalk, Cell Reports (2024). DOI: 10.1016/j.celrep.2023.113662
Journal information: Cell Reports
Provided by Emory University







Post comments