
by Christine Yu, Pennsylvania State University
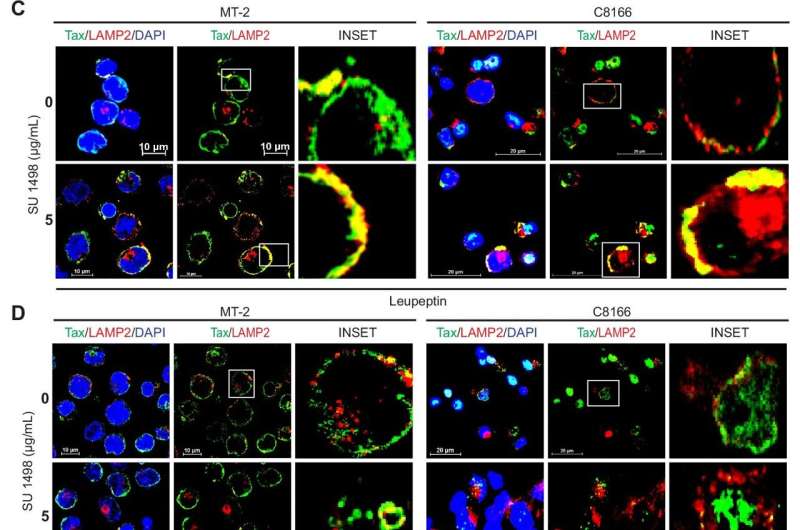
KDR inhibition induces autophagic/lysosomal degradation of Tax. Credit: Nature Communications (2024). DOI: 10.1038/s41467-024-49737-5
A team of researchers from Penn State College of Medicine found a new target for treating diseases associated with human T-cell leukemia virus type 1 (HTLV-1). They determined that blocking a class of enzymes called kinases, which regulate cellular functions, leads to cell death caused by the degradation of Tax, a protein essential for viral gene expression, viral transmission and survival of cells infected by HTLV-1. The team published the findings inNature Communications.
HTLV-1 is a retrovirus—a type of virus that hijacks a cell by inserting a copy of its genetic material into the host cells' DNA—and infects between 10 to 20 million people worldwide, primarily in southern Japan, central Australia, sub-Saharan Africa, South America, the Caribbean and the Middle East. Approximately 10% of those infected will develop adult T-cell leukemia/lymphoma (ATLL) or a neuroinflammatory disease similar to multiple sclerosis called HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP).
"HTLV-1 is understudied and there is currently a lack of effective treatments for the diseases it causes," said Edward Harhaj, professor of microbiology and immunology at Penn State College of Medicine and senior author of the study. "Our study could lead to possible new clinical approaches to target the Tax protein in patients infected by HTLV-1."
The research team set out to identify kinases that HTLV-1-infected cells need to survive. Using human cells transformed by the virus, the researchers performed what's called a short hairpin RNA screen—a molecular analysis that allowed the team to inhibit the expression of over 600 genes that encode kinases, one-by-one.
The results showed that only KDR, a tyrosine kinase also known as VEGFR2, was essential for the viability of the cells. To validate their findings, the team then treated the cells with small-molecule inhibitors that target KDR, including one that is a Food and Drug Administration-approved tyrosine kinase inhibitor. When KDR was blocked, the cells died.
"KDR wasn't on our radar because it's normally expressed in endothelial cells and regulates blood vessel formation," Harhaj said. "We were surprised that it was expressed in T cells—a type of white blood cell that protects against infection—and this particular leukemia we were studying. No one has ever implicated it before for the survival of these particular cells."
The study showed that KDR's role in the survival of HTLV-1-infected cells is connected to the viral protein called Tax. Tax is crucial for viral gene expression, viral transmission and the development of cancer. Suppression of KDR leads to the degradation of Tax and disrupts cancer-causing signaling pathways, leading to cell death. Cells that didn't express Tax weren't sensitive to KDR inhibition and didn't die. The team saw the same results when they inhibited KDR in blood samples from patients with HAM/TSP.
"We've been studying the Tax protein for a long time, but no one has found a way to target it. We found a potential way by targeting the host kinase KDR," Harhaj said. "KDR is not normally expressed in T cells, but Tax turns on its expression and hijacks its function, enabling it to stabilize and protect itself from degradation."
The findings point to a potential drug target for the treatment of ATLL and HAM/TSP. The researchers said that repurposing an existing KDR inhibitor or developing a new one could also potentially reduce the viral load of HTLV-1, possibly reducing the risk of developing disease.
"Clinically, KDR inhibitors could be very impactful, either by treating patients with disease or giving it to individuals with high viral loads to prevent disease," Harhaj said.
The team said they plan to continue this line of research.
More information: Suchitra Mohanty et al, The tyrosine kinase KDR is essential for the survival of HTLV-1-infected T cells by stabilizing the Tax oncoprotein, Nature Communications (2024). DOI: 10.1038/s41467-024-49737-5
Journal information: Nature Communications
Provided by Pennsylvania State University




Post comments