
by Bob Yirka , Medical Xpress
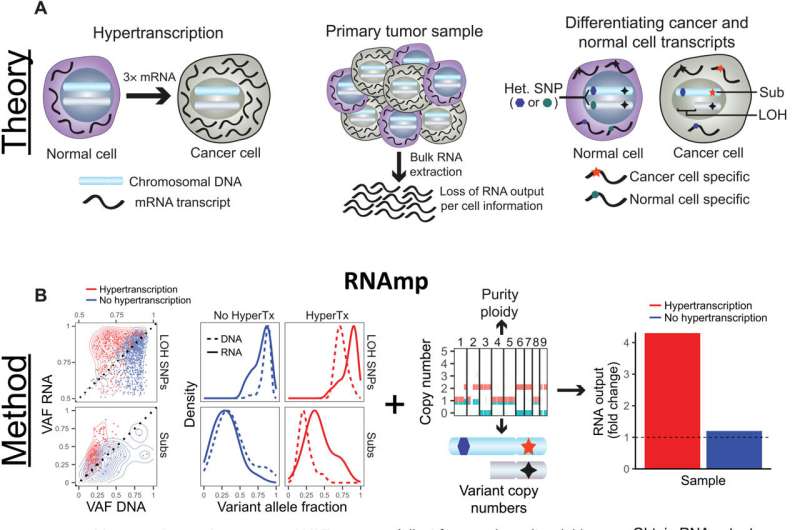
Overview of RNA output analysis with RNAmp. (A) Hypertranscription occurs when cancer cells elevate their RNA output above normal cell levels (left). Upon RNA extraction from primary tumor tissue, RNA output per cell information is lost (middle). Cancer cell—and normal cell—specific transcripts can be identified using tumor-specific marker variants, such as somatic substitutions (Subs) and LOH-SNPs (right). (B) DNA and RNA VAF distributions in samples with and without hypertranscription (HyperTX). Positive shifts in the RNA VAF of tumor-specific variants indicate that RNA output has increased. To estimate the overall fold change in RNA output of cancer versus normal cells, RNAmp incorporates these VAF shifts with tumor purity, ploidy, and local copy number data. (C) Cell number–normalized RNA-seq was performed on tumor and normal cell mixtures to validate RNAmp’s accuracy. RNA output per cell was measured before cell mixing. These mixtures were then sequenced and processed by RNAmp. (D) Fold change in RNA output levels of cancer cell lines measured by direct RNA quantification. Error bars correspond to SD. (E) RNAmp-derived RNA output measures (boxplots) compared to direct RNA quantification measures (red diamonds). Boxplot center line corresponds to the median, box limits are upper and lower quartiles, and whiskers represent 1.5 × interquartile range. (F) Pearson correlation of RNAmp-derived tumor RNA content estimates compared to direct RNA content quantification (R = 0.99, P < 0.0001). (G) RNA output per cell measured in medulloblastoma cells with and without MYC induction. (H) RNAmp-derived fold change in RNA output between UW228 Myc and UW228 wild-type cells (boxplot) compared to direct RNA quantification (red line). Boxplots are defined in (E). Credit: Science Advances (2022). DOI: 10.1126/sciadv.abn0238
A team of researchers from the University of Toronto, The Hospital for Sick Children, Toronto, the Ontario Institute for Cancer Research, Georgetown University and the Memorial Sloan Kettering Cancer Center, used a computational method to directly measure tumors, showing that hypertranscription is ubiquitous across cancers.
In their paper published in the journal Science Advances, the group describes developing a new computational tool to measure levels of transcription across all of the genes in a tumor and then used it to learn more about the impact of hypertranscription on survivability of cancer patients.
Prior research has shown that RNA is synthesized from DNA in a process called transcription, resulting in a production of a gene product such as a protein. Prior research has also shown that as tumors begin to grow, transcription generally goes awry, as well, leading to hypertranscription.
Medical researchers have wondered for some time whether some or all of the cells in a tumor have alterations to transcriptions, but have been unable to find out due to technical challenges.
In this new effort, the researchers overcame these technical challenges by developing a new tool that they named RNAmp. When trained on tumor mutation data, tumor type and other data such as changes to chromosomal structure, it returns with a normalized RNA measure for the entire tumor.
With their new tool in hand, the researchers set out to answer the question of whether hypertranscription occurs only in select cells or all of the cells throughout a tumor. They studied transcription levels in 7,494 tumors covering 31 kinds of cancer and found that hypertranscription was pervasive throughout a given tumor, regardless of type. Levels of transcription, they found, were universally high in all of the tumors they tested.
The researchers also found that the degree of hypertranscription could be associated with survivability—higher levels of transcription were correlated directly with shorter survival times. The researchers were not able to find a reason for the correlation, but note that use of their new tool when diagnosing patients could provide patients with more accurate estimates of their survivability time frame.
On a more positive note, this new finding suggests that future research focused on reducing hypertranscription in tumors could lead to better outcomes.
More information: Matthew Zatzman et al, Widespread hypertranscription in aggressive human cancers, Science Advances (2022). DOI: 10.1126/sciadv.abn0238
Journal information: Science Advances
© 2022 Science X Network



Post comments