
by Higher Education Press
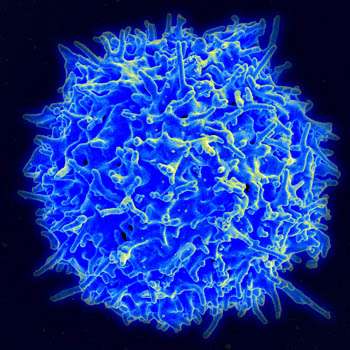
Scanning electron micrograph of human T lymphocyte or T cell. Credit: NIAID/NIH
In recent years, cancer immunotherapies, represented by immune checkpoint blockade (ICB), have been highly successful and have become an important basis for the future treatment of cancers. However, the absence of tumoral killer T cells and the complexity of tumor microenvironment can both affect the immunotherapeutic efficacy. Therefore, it is urgent to develop novel anti-tumor agents that can effectively promote effector T cell infiltration in tumors.
ADP-ribosylation factor 1 (Arf1) is a member of the Ras small GTPase family and is involved in regulating many essential physiological processes. However, several studies have shown that Arf1 is highly expressed in many human cancers, such as hepatocellular carcinoma, colon cancer, and breast cancer. Therefore, Arf1 is a potential target for tumor therapy. However, current inhibitors targeting Arf1 are generally characterized by high toxicity and poor specificity. Thus, it is necessary to develop novel Arf1 inhibitors that are safer and more effective for tumor treatment.
Researchers from the Department of Cell and Developmental Biology at School of Life Sciences of Fudan University developed two designed Arf1 inhibitors and elucidated their anti-tumor mechanism.
Their work focused on how blocking Arf1 promoted the infiltration of cytotoxic T lymphocytes into tumors by affecting lipid metabolism. This will provide a new therapeutic option for cancer patients and will improve the clinical outcome of cancer immunotherapy. This study titled "Blockade of Arf1-mediated lipid metabolism in cancers promotes tumor infiltration of cytotoxic T cells via the LPE-PPARγ-NF-κB-CCL5 pathway" was published online in Life Metabolism.
First, the two novel Arf1 inhibitors, which were previously developed by the team, could both significantly increase T cell infiltration within tumors. Tissue RNA sequencing data revealed a strong association between chemokines and T cells. By detecting the expression level of multiple chemokines and performing the tumor-immune cell co-culture assay, they found that Arf1 inhibition mainly influenced T cell migration through upregulating the level of chemokine CCL5 in tumor cells.
Next, in order to investigate how Arf1 blockade interfered with CCL5 transcription, they performed a correlation analysis of RNA-seq data from hepatocellular carcinoma patients in the TCGA database.
They found that the peroxisome proliferator-activated receptor gamma (PPARγ) signaling pathway was less activated in tumor tissues compared to normal tissues. PPARγ is a transcriptional regulator that can be activated by unsaturated fatty acids. Through lipidomic analysis, the researchers found that blocking Arf1 upregulated the level of polyunsaturated fatty acids (SUFA) and phosphatidylethanolamine (PE 18:1), especially lyso-phosphatidylethanolamine (LPE).
Further results showed that LPE indeed increased the expression of downstream genes in the PPARγ signaling pathway, suggesting that inhibition of Arf1 may affect the activation of PPARγ through the unsaturated fatty acid LPE. Previous study found that nuclear factor kappa B (NF-κB) is a major transcriptional factor in CCL5.
The team then analyzed whether NF-κB was involved in the transcriptional regulation of CCL5. According to the results of western blot and immunohistochemistry (IHC), they observed that Arf1 inhibition increased the expression level of phosphorylated p65.
The ChIP-qPCR assay showed that the binding ability of the NF-κB protein to the CCL5 promoter region was also significantly enhanced. However, what was the relationship between NF-κB and PPARγ activated by the unsaturated fatty acid LPE? Co-immunoprecipitation (Co-IP) experiment revealed that LPE attenuated the direct binding of PPARγ and NF-κB, suggesting the interference effect of Arf1 in their interaction.
To further investigate whether PPARγ-NF-κB affected CCL5 transcription, they performed electrophoretic mobility shift assay (EMSA) and found that the migrating band only appeared in the lane co-incubated with the CCL5 probe and p65 protein. In contrast, when the CCL5 probe was incubated with the PPARγ protein and p65 protein together, no migrating band was detected. They speculated that the PPARγ protein and CCL5 probe may compete for binding to the p65 protein, which may demonstrate the relationship between PPARγ-NF-κB-CCL5.
This study first figured out the mechanism of how blocking Arf1 promoted the infiltration of cytotoxic T lymphocytes into tumors by affecting lipid metabolism. Researchers demonstrated that Arf1 inhibition induced the formation of unsaturated fatty acids (LPE) which bound PPARγ and "snatched" PPARγ from the cytoplasmic PPARγ-NF-κB complex.
The "released" NF-κB was phosphorylated and translocated to the nucleus, where it regulated the transcription of chemokine CCL5. CCL5 bound to the CCR5 receptor on the surface of cytotoxic T cells, which further mediated T cell infiltration and ultimately regressed tumors.
Therefore, the regulation of the LPE-PPARγ-NF-κB-CCL5 axis by blocking Arf1 effectively promoted the migration of killer T cells into tumors and exerted anti-tumor effects. The team's findings provide a new treatment option for cancer patients exhibiting elevated Arf1 expression, offering hope for enhancing clinical outcomes in cancer care.
More information: Na Wang et al, Blockade of Arf1-mediated lipid metabolism in cancers promotes tumor infiltration of cytotoxic T cells via the LPE-PPARγ-NF-κB-CCL5 pathway, Life Metabolism (2023). DOI: 10.1093/lifemeta/load036
Provided by Higher Education Press





Post comments