
by Columbia University School of Engineering and Applied Science
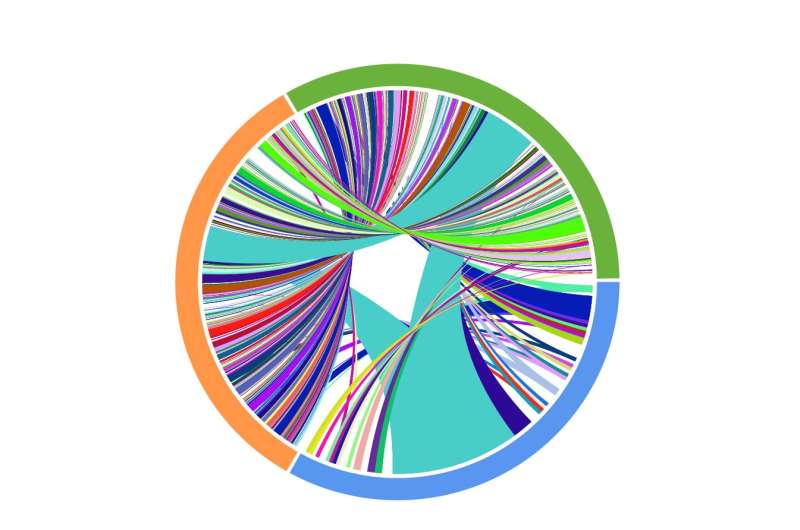
A Circos plot representing the results of matched T-cell receptor sequencing collected on a patient undergoing anti-PD1 therapy. Colors indicate time of collection (blue: pre-treatment, orange: on treatment, green: on-later), and connections indicate overlap of identical TCRs. These results demonstrate a striking diversification of T-cell clonotypes over time. Credit: Yiping Wang/CUIMC
Researchers at Columbia Engineering and Columbia University Irving Medical Center have invented a new RNA sequencing method that achieves high-quality results from small volumes of frozen tumor specimens. They demonstrated the success of their technique in two clinical studies that profiled dozens of tumor samples, both those archived and those freshly collected, to understand how they respond to anti-tumor therapy. The paper was published January 9, 2023, by Nature Genetics.
Using RNA sequencing to measure gene expression at the resolution of single cells has been one of the most transformative tools for studying cancer tissues over the past decade. By examining the RNA of individual cells, researchers can better understand the diversity of cells within a tumor, as well as how these tumor cells grow and interact with immune cells. These are important factors for understanding the hallmarks of cancer progression and the resistance of cancer to therapy—both key to the development of new cancer treatments.
A major barrier to the widespread adoption of single-cell RNA sequencing in traditional clinical workflows has been the volume of fresh tissue required—substantially more than what is routinely collected for clinical purposes. And the need for fresh tissue means that samples must be immediately analyzed once they are collected. These requirements have significantly limited the scientific analyses that can be done on patient samples.
Successful new technique
Focused on overcoming these barriers, the Columbia team created a new sequencing method that delivered top results from a small amount of frozen tumor specimens. Usually collected during clinical trials and stored in biobanks, these specimens may include tissues from rare cancers and patients with unique histories or risk factors. The new technique's ability to sequence these types of specimens greatly increases the number and variety of tumor samples available for scientific analysis.
"We are very excited about this work and its potential," said Elham Azizi, assistant professor of biomedical engineering and Herbert and Florence Irving Assistant Professor of Cancer Data Research at the Irving Institute for Cancer Dynamics, who co-supervised this study with Benjamin Izar, assistant professor of medicine at the Herbert Irving Comprehensive Cancer Center.
"The ability to work from frozen samples opens the door to multi-institutional collaborations that will propel the discovery of biomarkers and drug targets. It also gives us the opportunity to apply cutting-edge computational techniques in the analysis and integration of the unlocked clinical data," Azizi explained.
Izar added, "And because our method requires only a minute amount of tissue, the remainder of the sample may be used for additional studies. This really is a win-win for researchers, clinicians, and, most importantly, our patients."
Study's results broaden understanding of cancer progression
The new study led by Yiping Wang, Joy Fan, and Johannes Melms, trainees in the Izar and Azizi Labs, generated results from single-cell RNA sequencing, single-cell T-cell receptor sequencing, whole genome sequencing, and spatial RNA-sequencing (an innovative RNA-sequencing method that preserves the tumor architecture in situ)—all performed on the same samples.
With the ability to bridge multiple modalities of cancer data, the researchers provided a comprehensive view of the genetic alterations, cellular functions, immune cell dynamics, and spatial localization of cells in the context of the patient tissue. These improvements significantly broadened their understanding of cancer progression and resistance mechanisms.
The team is now applying their novel experimental and computational techniques to analyze larger clinical cohorts. The data enables them to better study disease progression and examine the impact of therapies in clinical trials in melanoma as well as other cancer types.
ECHIDNA, a new computational tool
In parallel, the researchers are also developing an innovative computational tool for systematic integrative analysis of whole genome data and single-cell RNA sequencing, named ECHIDNA. They expect this machine learning algorithm to be key in understanding the relationship between genetic alterations (genotype) and cellular function (phenotype) in tumor cells and will apply it to a larger cohort of melanoma patients in order to characterize diverse mechanisms of treatment resistance. Future work will also include building new computational methods for integrating these data within the spatial context of the histology slice in order to characterize the spatial dynamics of tumor-immune cell interactions.
Izar noted, "In addition to understanding cancer tissue, our method also enables studying tissue response and tissue immunology in other diseases. In the past, we used an earlier version of the method presented here to study the tissue response to lethal COVID-19 infection across multiple organs. This demonstrates an example of developments originating in cancer research that can propel life science research at large."
More information: Yiping Wang et al, Multimodal single-cell and whole-genome sequencing of small, frozen clinical specimens, Nature Genetics (2023). DOI: 10.1038/s41588-022-01268-9
Journal information: Nature Genetics
Provided by Columbia University School of Engineering and Applied Science






Post comments